

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor:
Dyschromatosis universalis hereditaria (DUH) is a rare genodermatosis characterized by hypo- and hyper-pigmented macules with a reticulated pattern, giving an overall impression of mottling, over the trunk and limbs. It is generally an autosomal dominant disorder but recessive inheritance and patients without a family history of dyschromatosis have been reported, implying a sporadic origin12. The etiology of this disorder is unknown. It was recently suggested to be a disorder of melanosome synthesis rate or melanocyte activity and not a disorder of melanocyte number3.
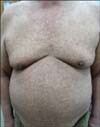
A 57-year-old man presented with generalized hypo- and hyper-pigmented macules in mottled pattern on the whole body (Fig. 1). The skin lesions developed on 100th day after his birth. He had moderate pruritus and dry skin with scaling. Potassium hydroxide preparation from the scale produced a negative result. The hair, nails, and oral mucosa as well as mental and developmental status were normal. He had no family history of similar lesions, but his son was diagnosed with X-linked ichthyosis at birth. The patient had a history of diabetes mellitus and hypertension. Systemic examination revealed no abnormalities. Routine blood test results were unremarkable. Skin biopsy specimens taken from both hypo- and hyper-pigmented lesions on his arms revealed markedly reduced and slightly increased melanin in the basal layer in hypoand hyper-pigmented lesions, respectively (Fig. 2). Therefore, we diagnosed the skin lesions as a sporadic form of dyschromatosis universalis.
Dyschromatosis is variation of skin pigmentation, consisting of asymptomatic well-demarcated and irregular brown macules admixed with hypo-pigmented macules of varying size2. There are three major categories of dyschromatosis: DUH, dyschromatosis symmetrica hereditaria (also termed acropigmentation of Dohi), and unilateral dermatomal pigmentary dermatosis. Since its first description by Toyamo in Japan in 1929, subsequent cases of DUH have been reported in Europe, South America, and India. The phenotype seems to be heterogeneous. It is characterized by the presence of both hyper- and hypopigmented small irregular macules uniformly distributed all over the body. The trunk and extremities are the predominantly affected sites, while the face, palms, soles, and mucous membranes are less affected. The skin lesions usually develop in the first few months after birth and do not progress with age. Histopathologically, hyperpigmented lesions exhibit increased melanin content in the basal layer as well as melanin incontinence, while hypopigmented lesions exhibit relatively decreased melanin deposition in the basal layer2. On electron microscopy, hyperchromic macules contain numerous fully melanized melanosomes forming melanosome complexes, but melanosomes are absent from both keratinocytes and melanocytes in achromic macules3. The pathogenesis of DUH remains unknown. However, the DUH locus was recently mapped to chromosome 6q24.2-q25.2 and 12q21-q23, and the ABCB6 gene was identified as the pathogenic gene4.
Most cases of DUH have been reported in Japan while only a few have been reported in Korea5. Furthermore, the sporadic form has rarely been reported in the Korean literature. In summary, we report a rare case of the sporadic form of dyschromatosis universalis in a middle-aged man who had no family history.

 XML Download
XML Download