

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor:
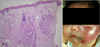
A 15-day-old boy visited our clinic presenting a palm sized erythematous patch located on his left cheek from birth, which was aggravated soon after his 1st vaccination against the hepatitis B virus. A skin biopsy revealed an oval shaped vascular lobules scattered in the dermis. It was a typical 'Cannonball' appearance (Fig. 1A). In high power field, the lobules were groups of ducts composed of proliferated endothelial cells. Tufted angioma (TA) was the most consistent diagnosis. The patient began receiving interferon a2b injection of 150 MU four times a week. However, the injection was ceased because 18 injections during a course of five weeks only brought intermittent fever with minimal effect. As an alternative option, propranolol (15 mg/d) and prednisolone (3 mg/kg/d) were given to the patient. Although a short-term tumor shrinkage was achieved, intermittent flare-ups of swelling, pain and tenderness persisted (Fig. 1B). Even vincristine (0.05 mg/kg/wk) failed to downsize the tumor.
The laboratory result at the four-month-follow up revealed a decreased platelet (107,000/ul), elevated D-dimer (4.66 ug/ml) and decreased fibrinogen (136 mg/dl), which implied the advent of Kasabach-Merritt syndrome (KMS). On this, he underwent nine sessions of combined chemotherapy of vincristine 0.05 mg/kg, actinomycin-D 1.5 mg/m2, cyclophosphamide 500 mg/m2 until now. For the one-year follow up since his first visit, neither the size of the tumor nor the KMS satisfactorily responded to the various treatment modalities. He still suffers from outbreaks of edema, tenderness and thrombocytopenia. For acute aggravation, methylprednisolone pulse therapy (30 mg/kg/day for 3 consequent days) and platelet transfusion were conducted for edema and thrombocytopenia, respectively (Fig. 2).
TA is a rare benign vascular tumor, which originates from the abnormal proliferation of endothelial cells1. Another vascular tumor, Kaposiform Hemangioendothelioma (KHE), shares many common properties with TA. Both have similar histopathology and can cause KMS. Although the immunohistochemistry for D2-40 differentiates KHE (positive for this marker) and TA (negative), it has currently been suggested that both are spectrums of the same vascular tumor. Unfortunately, there are no established treatments. Only several cases of using systemic agents or laser have been reported. It was suggested that physical trauma possibly aggravates the tumor by introducing various growth factors2. This hypothesis partly explains several cases of aggravated TA after skin punch biopsy at the authors' clinic (unpublished observation). We also experienced an aggravation of TA followed by a long pulsed Nd:YAG laser3. Interestingly in this case, the aggravation took place along with fever after vaccination. If the aggravation was correlated with mild inflammatory response caused by vaccination, we could hypothesize that even a mild inflammation triggers the proliferation of vascular tumor cells. Our experiences imply that TA is also associated with Koebner phenomenon (KP), which is similar to another vascular tumor, Kaposi's sarcoma, which is known to be developed as a manifestation of KP4.
KMS is a rare condition characterized by thrombocytopenia and consumptive coagulopathy, arising in patients with vascular tumor. Treatment experiences using corticosteroid and chemotherapeutic agents have been reported5.

 XML Download
XML Download