

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Light chain deposition disease (LCDD) is a systemic disease characterized by clonal proliferation of plasma cells, overproduction of abnormal light chains, and deposition of non-amyloid monoclonal immunoglobulin light chains in various organs, which can induce organ dysfunction, especially in the kidney (1, 2). This disorder was originally described by Randall et al. (5) in 1976 in two patients with end-stage renal disease (ESRD) with granular deposition of free light chains that did not stain with Congo red on kidney pathologic evaluation. There have only been a few reports of isolated LCDD involving the brain (3), lungs (4), cervical lymph nodes (5), and pharynx (6). Here, we report one case of LCDD limited to the duodenal mucosa. Based on laboratory and radiologic tests, especially normal serum and urine-free light chain levels, we conclude that no systemic signs of the disease were present in this patient at any time point in the disease process.
CASE DESCRIPTION
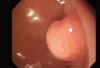
A 63-yr-old man visited Chung-Ang University Hospital for a regular examination without any symptoms or complaints on March 1st, 2009. An esophagogastroduodenoscopy revealed a polypoid mass in the duodenal bulb measuring approximately 2 cm (Fig. 1). An endoscopic biopsy from the mass showed a deposition of pink, amorphous material and infiltration of plasma cells in the lamina propria (Fig. 2A). Congo red staining was negative (Fig. 2B). A few plasma cells showed λ light chain (LC) immunohistochemical staining (Fig. 2C), although most plasma cells were κ light chain-positive (Fig. 2D).
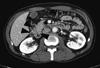
Following the biopsy results, a complete skeletal X-ray survey, abdomino-pelvic computed tomography (CT), bone marrow histology (including karyotyping and serum/urine-free LC levels), serum/urine immunofixation electrophoresis, and serum/urine protein electrophoresis were performed. No abdominal mass was palpated on physical examination. The CT scan showed multiple inhomogeneous polypoid masses in the duodenum (Fig. 3). Lymphadenopathy was not detected. A serum-free LC assay revealed slight elevation of kappa chains at 28.4 mg/L (normal range, 3.3-19.4 mg/L), while lambda chains were normal at 25.2 mg/L (normal range, 5.71-26.3 mg/L) with a normal λ:k ratio of 1.13 (normal range, 0.26-1.65). A mild predominance of serum kappa chains was consistent with the kappa stained-positive duodenal polyp. Serum beta-2-microglobulin was elevated at 2.43 mg/L (normal range, 0.81-2.19 mg/L). A subsequent work-up (serum/urine protein electrophoresis, radiographic examination of the axial skeleton, and a bone marrow aspirate and biopsy) was completely negative. A bone marrow aspirate and biopsy revealed 1.2% plasma cells and normal cytogenetics. Laboratory testing was normal, as follows: white blood cell count, 5,880/µL; hemoglobin, 15.4 g/dL; platelet, 212,000/µL; blood urea nitrogen, 13 mg/dL; creatinine, 1.1 mg/dL; total protein, 6.7 g/dL; albumin, 3.7 g/dL; and 24-hr-urine protein, 1.6 g/dL. On the basis of these features, localized LCDD in the duodenum was diagnosed. The patient refused aggressive treatment or regular follow-up. Thereafter, any polyps around the duodenum were not observed in arbitrarily applicated esophagogastroduodenoscopy on May 5th, 2011.
DISCUSSION
LCDD is a rare clinicopathologic entity characterized by tissue deposition of non-amyloid immunoglobulin light chains (7). LCDD affects middle-aged patients ranging from 35-76 yr, with a mean of 56 yr. LCDD affects men 2.5 times more often than women (1). LCDD is usually associated with monoclonal gammopathies of undetermined significance in 17% of patients and MM in 58% of patients (8). However, there were no reports of LCDD involving a duodenal polyp. Unlike AL amyloidosis, one of the monoclonal light chain deposition diseases in tissues, LCDD does not stain with Congo red, which displays green birefringence under polarization.
Clinical manifestations of LCDD are related to the underlying condition and the location of deposits. Renal involvement is consistently present and is characterized by proteinuria and microscopic hematuria. In most patients with LCDD, renal function declines rapidly as a rapidly progressive glomerulonephritis (1) or as an acute tubulointerstital nephritis (9), which is due to progressive accumulation of light chains from plasma filtration and includes proteinuria, nephrotic syndrome, and/or renal failure. Symptomatic extrarenal deposition is rare and has been described in the heart (10, 11), liver (12), lungs (1, 9, 10), joints (11), and central and peripheral nervous systems (3). It is uncertain whether or not localized LCDD really exists or represents an initial expression of a silent systemic LCDD (5).
Among nonamyloid monoclonal immunoglobulin deposition disorders, the most frequent form of is κ light chain deposition disease (13, 14), which was also the diagnosis of this case. Lamda light chain deposition represents 15%-20%, and combined light and heavy chain deposition fewer than 10% of the reported cases. As the clinical presentation in LCDD is known to depend on the number and nature of the organs affected, deposition of different light chains does not seem to affect their clinical course.
The median duration of survival in systemic LCDD is approximately 4 yr. Prognostic factors for LCDD include age, presence of plasma cell myeloma, and extrarenal light chain deposition (9). The treatment of LCDD has not been established. Chemotherapy with steroids and melphalan has been used, but the response rates have been low (15). A combination of high-dose melphalan and autologous stem cell transplantation has been reported to improve renal function in affected patients (16), but high mortality rates and side effects limit this regimen (17).
In conclusion, we have reported the first case of LCDD involving the duodenum alone in an asymptomatic patient. A less aggressive clinical course was expected in the patient presented herein compared to patients with systemic involvement of LCDD, but long-term follow-up is necessary to establish the clinical significance of isolated LCDD.

 XML Download
XML Download