

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Thyroid-stimulating hormone (TSH), insulin, and insulin like growth factor-1 (IGF-I) stimulate cell cycle progression and proliferation in various thyrocyte culture systems, including established rat thyroid cell lines (FRTL-5, WRT, and PC Cl3) and primary cultures of rat, dog, sheep, and human thyroid (1). TSH regulates thyrocyte proliferation via activation of cAMP-dependent and cAMP-independent signaling pathways, which involve protein kinase A, phosphatidylinositol 3 (PI3)-kinase, and RAS (1-5). TSH is not only involved in the proliferation of thyroid cells, but is also associated with the maintenance of differentiated functions, such as regulation of thyroglobulin and thyroperoxidase gene expression (6-9). The signaling pathways activated by TSH are associated with the induction and activation of a number of thyroid-specific and ubiquitous transcription factors, which transcribe a set of genes that control thyrocyte proliferation and maintenance of function. Specifically, TSH (5-9) and insulin (8-11) regulate thyroid-specific transcription factors, such as TTF-1, TTF-2, and Pax-8, and these transcription factors are critically involved in the transcriptional regulation of thyroglobulin (Tg), thyroperoxidase (TPO), TSH receptor (TSHR) (7, 8, 10), and the sodium iodide symporter (12, 13).
Inhibitors of differentiation (Id) proteins are small helix-loop-helix proteins that lack a DNA binding region. Usually these proteins interact with basic helix-loop-helix (bHLH) proteins and act as negative regulators by preventing their DNA binding and transcriptional activities (14). Four mammalian members of the Id protein that have been identified to date exhibit 69-78% identity at the amino acid level within their HLH dimerization domains, but other regions of these proteins are essentially unrelated (15). Id family proteins are important in the regulation of various cellular processes, including cellular proliferation, death, and differentiation (14, 16-24). Recent studies have revealed that Id proteins bind to pRB (retinoblastoma tumor suppressor protein) family proteins and Ets-family transcription factors, which are known to play key roles in cell cycle regulation, transformation, and tumor suppression (20-23).
Although the expression of Id family proteins is elevated in proliferating cells and down-regulated during differentiation, the precise pattern of their regulation is highly dependent on the cell type (22-25). In this report, we have investigated Id family protein expression in FRTL-5 thyroid cells following exposure to TSH and have demonstrated the effects of Id family proteins on the transcriptional activities of the thyroid-specific transcriptional factors, TTF-1 and Pax-8.
MATERIALS AND METHODS
Materials
Highly purified bovine TSH (Sigma Chemical Co., St. Louis, MO, U.S.A.), [γ-32P] dCTP (3000 Ci/mM) (DuPont-Merck Pharmaceutical Co., Wilmington, DE, U.S.A.), Wortmannin, LY294002 and H89 (Calbiochem, La Jolla, CA, U.S.A.), Rapamycin (Sigma Chemical Co.), and PD98059 (New England Biolabs, Beverly, MA, U.S.A.) were obtained. All other materials were from Sigma Chemical Co., unless otherwise noted.
Cells
FRTL-5 rat thyroid cells (Interthyroid Research Foundation, Baltimore, MD, U.S.A.) were a fresh subclone (F1) that had all properties detailed previously (26). Their doubling time in the presence of TSH was 36±6 hr, and in the absence of TSH they did not proliferate. After 6 days in medium lacking TSH, the addition of 1×10-10 M TSH stimulated thymidine incorporation into DNA by at least 10-fold. Cells were diploid and between their 5th and 20th passage. They were grown in 6H medium consisting of Coon's modified F12 supplemented with 5% calf serum, 1 mM nonessential amino acids, and a mixture of six hormones (6H): bovine TSH (1 mU/mL), insulin (10 µg/mL), cortisol (0.4 ng/mL), transferrin (5 µg/mL), glycyl-L-histidyl-L-lysine acetate (10 ng/mL), and somatostatin (10 ng/mL). The medium was replaced every 2 or 3 days and cells were passaged every 7-10 days. In individual experiments, cells were shifted to 0H medium (devoid of 6 hormones described above) and 5% calf serum. COS7 kidney fibroblasts were maintained in Dulbecco's modified Eagle's medium (high glucose) supplemented with penicillin-streptomycin and 10% fetal bovine serum (FBS) (GIBCO-BRL, Carlsbad, CA, U.S.A.).
Plasmids
The promoter reporter construct of the Id2 gene, pGId2-2750, was supplied by Dr. Lavarone A (22). The mutant Id2 promoter construct, pGId2-2750 del (EcoR1), was made by deletion of the EcoR1 fragment (1,330-2,230 bp) that contains high-affinity Myc-binding sites. GAL4-Id fusion constructs were generated by cloning cDNAs of Id1, Id2, Id3, and Id4 into pBind. The thyroglobulin promoter constructs, -808 Tg-pGL3, -688 Tg-pGL3, and -207 Tg-pGL3 were supplied by Dr. S. Kimura. The expression vectors, pRc/CMV-TTF1 and pRc/CMV-Pax-8 (7), were kindly provided by Dr. Kohn L.D. All plasmids were prepared using a Plasmid Maxi Kit (QIAGEN, Chatsworth, CA, U.S.A.).
Northern blotting
Total RNA was extracted from FRTL-5 rat thyroid cells cultured in a 10-cm dish following exposure to TSH or insulin as described previously (27). Approximately 20 µg total RNA was fractionated on a 1% formaldehyde-agarose gel. After fractionation, the RNA was transferred to a nylon membrane (Hybond+ N, Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.) in 10×SSC buffer and UV-cross-linked as described previously (27). The membranes were then prehybridized in Quick Hybridization buffer (Stratagene, La Jolla, CA, U.S.A.) for 1 hr at 69℃, following which the hybridization was carried out at 69℃ for 24 hr with 32P-labeled Id probes. Membranes were washed for 15 min at room temperature in wash buffer A (2X SSC and 0.1% sodium dodecyl sulfate [SDS]), and for 15 min at higher stringency temperature in wash buffer B (0.1×SSC and 0.1% SDS). All probes were radiolabeled using a random priming protocol (Amersham Pharmacia Biotech). The labeled probes used were the full length cDNA for rat Id1, Id2, Id3, Id4, and β-actin.
Immunoblot analysis
Immunoblot analysis was performed using anti-Id (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) antibodies. Adherent FRTL-5 cells were stimulated with various agents for the indicated period of time at 37℃. The treated cells were scraped, lysed by addition of SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 6% [w/v] SDS, 30% glycerol, 125 mM DTT, 0.03% [w/v] bromophenol blue) and separated by 10% SDS-PAGE, along with biotinylated molecular weight standards. The proteins were transferred to a nitrocellulose membrane by electrotransfer for 2 hr. After soaking in blocking buffer (1×TBS, 0.1% Tween-20, 5% milk) the membrane was incubated with primary antibody overnight at 4℃. Blots were developed using the HRP-linked secondary antibody and a chemiluminescent detection system (Phototope®-HRP Western Blot Detection Kit, New England Biolabs, Beverly, MA, U.S.A.).
Transient transfection and luciferase assay
Cells were transfected by the LipofectAMINE method (Invitrogen, Carlsbad, CA, U.S.A.) according to the manufacturer's instructions. Briefly, 400 ng of promoter reporter construct (Fig. 7A) was incubated in serum-free media with 6 µL LipofectAMINE Plus reagent at room temperature for 15 min; 4 µL LipofectAMINE reagent was added and the mixture was incubated at room temperature for a further 15 min. Semiconfluent cells were washed twice with 1× PBS (phosphate-buffered saline) and incubated with DNA-LipofectAMINE Plus reagent complexes at 37℃ in a humidified chamber containing 5% CO2 for 4 hr. All DNA constructs were transfected with 100 ng pRL-SV40 plasmid encoding Renilla luciferase (Promega, Madison, WI, U.S.A.) to adjust the transfection efficiency. Following transfection, cells were allowed to recover for 24 hr following which they were washed with 1×PBS and lysed with 100 µL lysis buffer containing 40 mM Tricine (pH 7.8), 50 mM NaCl, 2 mM EDTA, 1 mM MgSO4, 5 mM DTT, and 1% Triton X-100. Extracts were assayed in triplicate for luciferase activity in a total volume of 130 µL containing 30 µL cell extract, 20 mM tricine, 0.1 mM EDTA, 1 mM magnesium carbonate, 2.67 mM MgSO4, 33.3 mM DTT, 0.27 mM coenzyme A, 0.47 mM luciferin, and 0.53 mM ATP, and light intensity was measured using a luminometer (Berthold, Bad Wildbad, Germany). Firefly and Renilla luciferase activities were measured using a Dual-Luciferase Reporter assay system (Promega, Madison, WI, U.S.A.) and a model TD-20/20 Luminometer. Luciferase activity was integrated over a 10-sec period. Firefly luciferase values were standardized to Renilla values.
GAL4 hybrid assays
The pG5-luc reporter plasmid contains five GAL4-binding sites upstream of the coding sequence with a minimal thymidine kinase region for luciferase assay. The GAL4-fusion protein expression plasmids; pBind-Id1, pBind-Id2, pBind-Id3, and pBind-Id4 were coexpressed with the pG5-luc reporter in FRTL-5 thyroid cells. Transfected cells were treated with or without 1 mU TSH for 12-24 hr and assayed for luciferase activity as previously described (28).
RESULTS
Regulation of Id RNA expression by TSH and insulin in FRTL-5 cells
Expression of Id transcripts was observed in FRTL-5 thyroid cells following treatment with TSH (1 mU/mL) and insulin (10 ng/mL) (Fig. 1). Id2 and Id4 mRNA expression was increased by TSH (1 mU/mL) treatment; however Id1 and Id3 were not modulated. The induction of Id2 mRNA by TSH was rapid and prolonged. Its maximal level was reached within 1 hr and this level was maintained for 12 hr (Fig. 1A). Id4 mRNA was also rapidly induced (within 1 hr) by TSH treatment but returned to its basal level by 6 hr. Different patterns of Id RNA expression were observed in insulin-treated cells. The TSH-responsive Id2 did not respond to insulin, whereas Id1 and Id4 RNA increased following exposure to insulin, and Id3 mRNA expression was virtually unaffected.
Regulation of Id protein levels by TSH in FRTL-5 thyroid cells
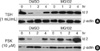
Since the Id RNA expression studies suggested that Id2 is a major TSH-responsive gene, we investigated Id2 protein levels in FRTL-5 cells following TSH treatment. Id2 protein was regulated biphasically in response to TSH treatment. As shown in Fig. 2A, Id2 was expressed basally in untreated FRTL-5 thyroid cells; however, the level of Id2 rapidly decreased within 2 hr of TSH treatment and subsequently recovered to the basal level. After an 8-hr exposure to TSH, the expression of Id2 was higher than that of untreated cells. The influence of TSH on FRTL-5 cell signaling was further monitored by detection of S6 ribosomal protein phosphorylation (4, 5). This phosphorylation event reflects the activity of S6 kinase, which is activated by both TSH and forskolin in thyroid cells (Fig. 2). Phosphorylation of ribosomal S6 protein was observed in TSH-treated cells within 1 hr and was maintained for 12 hr. In a similar experiment, forskolin induced a transient decrease of Id2 protein expression that coincided with the TSH-mediated activation of S6 kinase. Insulin also transiently decreased Id2 protein expression during the S6 kinase activation phase.
Previously, it has been shown that Id family proteins are short-lived proteins whose degradation is dependent on the proteosomal pathway (29). As shown in Fig. 2A, Id2 was rapidly down-regulated, which suggested that Id2 proteins would also be degraded by the proteasomal pathway in response to TSH treatment. To investigate this, we pretreated FRTL-5 rat thyroid cells with or without MG132, a specific proteosome inhibitor (30, 31), prior to TSH treatment. MG132 completely blocked Id2 degradation induced by TSH or forskolin treatment (Fig. 3). These results strongly support the hypothesis that TSH- and forskolin-induced Id2 degradation is mediated by the proteosomal pathway.
The phosphatidylinositol 3 kinase pathway is involved in TSH-mediated Id2 degradation
Given that the transient decrease of Id2 level in TSH-treated cells occurred during the activation phase of S6 kinase, we hypothesized that PI3 kinase may be involved in TSH-mediated Id2 degradation in thyroid cells. Moreover, activation of S6 kinase by TSH is dependent on PI3 kinase, mTOR, and PKA in thyroid cells (1-5). To identify the involvement of PI3 kinase, mTOR and PKA signaling in TSH-induced Id2 degradation, the cells were pretreated with the specific inhibitors LY294002, wortmannin, rapamycin, H89, and PD98059 (3, 4) for 2 hr prior to incubation with TSH or forskolin. Both LY294002 and wortmannin, which are specific PI3 kinase inhibitors, prevented TSH-mediated Id2 degradation (Fig. 4A, B). However, rapamycin, a specific inhibitor of mTOR, and H89, a PKA inhibitor, had no effect on Id2 degradation (Fig. 4C). PD98059, a MAPK inhibitor, also had no effect on TSH-induced Id2 degradation (data not shown). These results indicate that PI3 kinase pathways are involved in TSH-induced proteosomal degradation of Id2.
Regulation of Id2 promoter activities by TSH in FRTL-5 thyroid cells
In the above studies, we found that Id2 mRNA was induced by TSH treatment, and that its protein level was also upregulated after 8 hr by TSH treatment. To confirm if Id2 promoter activity is regulated by TSH, we transfected FRTL-5 cells with pGId2-2750, a luciferase reporter that contained-2,750 bp of the 5'-region of the mouse Id2 gene (22) and measured its activity following TSH treatment. The high affinity Myc-binding sites (CACATG at position -1,880 and CACGTG at position -1,950) in the Id2 promoter are shown in Fig. 5A. These E-boxes are known to be important sites for Myc-mediated transactivation of the Id2 promoter during serum-mediated fibroblast stimulation (22). The construct, pGId2-2750 del (1330-2230), did not contain the Myc-binding sites. TSH increased not only wild-type pGId2-2750 promoter activities, but also increased the activity of the mutant pGId2-2750 del (1330-2230) that is devoid of Myc-binding sites. Although several studies suggested that TSH can induce c-Myc expression in thyroid cells, these results suggest that induction of Id2 promoter activities in FRTL-5 thyroid cells by TSH may not depend on c-Myc.
Regulation of transcriptional activities of Id proteins and effects of Id2 protein on TTF-1/Pax-8 activity in FRTL-5 cells
In the above study, we found that TSH regulates the induction of Id2 promoter activity, mRNA and protein expression in FRTL-5 thyroid cells. All four Id family proteins possess related HLH dimerization domains that can interact with similar bHLH proteins. Recent studies suggested that Id family proteins are phosphorylated and have transcriptional activities (32). We tested the effects of TSH on the transcriptional activities of Id proteins using GAL4-dependent transcriptional systems. GAL4 fusion protein expression plasmids, GAL4-Id1, GAL4-Id2, GAL4-Id3, and GAL4-Id4, were co-transfected with the reporter plasmid pG5-luc, and reporter activities were monitored in the presence or absence of 1 mU TSH. As shown in Fig. 6, GAL4-Id1, GAL4-Id3, and GAL4-Id4 proteins did not activate GAL4-dependent transcription. GAL4-Id2 protein had weak, albeit significant, transcriptional activity, but this was not modulated by TSH. These observations suggest that TSH signaling does not directly affect Id2-mediated transcriptional activity in FRTL-5 thyroid cells.
Id proteins have been shown to interact with Pax family transcription factors, such as Pax-2/-5/-8, and to inhibit the DNA binding of Pax transcription factors (24). Pax-8 is a thyroid-limited transcription factor, which is involved in normal thyroid development and transcriptional regulation of the thyroid-specific genes Tg, TPO, and sodium iodide symporter (NIS) (4-8, 13). To determine whether Id2 is involved in regulating transcription of the Tg gene, we cotransfected the Tg promoter constructs, -808 Tg-pGL3, -688 Tg-pGL3, and -207 Tg-pGL3, with or without the expression plasmid constructs pCDNA3-Id2 into FRTL-5 cells. As shown in Fig. 7A, Id2 did not change the reporter activities of -808 Tg pGL3, -688 Tg pGL3, or -207 Tg pGL3, which has binding motifs for Pax-8 and TTF-1. In addition, in Cos-7 cells, Id2 did not affect TTF-1-mediated or Pax-8-mediated Tg gene transcription (Fig. 7B, C). These observations suggest that Id overexpression may not alter the expression of thyroid-specific genes such as Tg in thyroid cells.
DISCUSSION
In the present study, we investigated the regulation of expression and functions of Id family proteins in FRTL-5 thyroid cells. We found that the expression of Id2, is up-regulated by TSH in FRTL-5 thyroid cells. At the protein level, the regulation of Id2 by TSH was biphasic, starting with a transient decrease followed by a delayed increase in expression, although Id2 mRNA was rapidly induced by TSH. The transient decrease of Id2 protein expression was not specific to TSH, but was also observed following insulin treatment. Moreover, the rapid degradation of Id2 by TSH was prevented by MG132, a specific proteosome inhibitor (30, 31). This observation suggests that TSH may modulate the activities of E1 ubiquitin enzyme systems.
PI3 kinase signaling pathways are involved in regulating differentiation in many cell types (4, 5, 33, 34) and are also involved in protein degradation (32, 34-36). For example, insulin-induced IRS-1 degradation requires the PI3 kinase pathway (31, 35, 36). As shown in Fig. 4, the specific PI3 kinase inhibitors, LY 294002 and wortmannin, prevented TSH-induced Id2 degradation in FRTL-5 cells (Fig. 4). Several studies have suggested that TSH activates PI3 kinase-dependent signaling pathways to regulate thyrocyte proliferation (1-5). We propose that PI3 kinase pathways that are activated by TSH may be responsible for the degradation of specific proteins, such as Id2, in thyroid cells. However, the biological significance of the transient reduction of Id2 in TSH-activated thyroid cells was not fully evaluated in this study.
The expression of Id2 is transcriptionally regulated through the activation of several transcription factors, such as c-Myc (22). The human Id2 promoter contains a cluster of three E-boxes that are high-affinity Myc-binding sites (CACATG at position -1,880 and CACGTG at position -1,590) (22). TSH and cAMP both enhance the expression of the c-Myc proto-oncogene in cultured thyroid cells (37). We further tested whether the Id2 promoter activation was under the direct control of c-Myc in FRTL-5 cells. However, the inducibility of a mutant Id2 promoter, which has no Myc-binding sites, by TSH was comparable to the wild-type Id2 promoter. These findings suggest that Id2 induction by TSH may not involve c-Myc-dependent transcriptional mechanisms.
TSH is known to regulate differentiation functions in thyroid cells (3-9, 12, 13). Specifically, TSH regulates the expression of thyroid-specific genes, such as Tg, TPO (6-10) and NIS (12, 13) by modulating thyroid transcription factors. Recent studies suggest that Pax-2/-5/-8 bind to Id proteins and that Id proteins antagonize the activity of members of the Pax transcription factor family (24). Thus, we investigated potential functional cross-talk between Id2 and thyroid-specific transcription factors in both thyroid and non-thyroid cells (Fig. 7). Id2 overexpression did not alter Tg promoter activity in thyroid cells and did not affect TTF-1 or Pax-8-induced Tg promoter activities. These overexpression experiments suggest that Id2 does not cross-talk with thyroid transcription factors in the regulation of Tg gene transcription. Because thyroid transcription factors, TTF-1 and Pax-8, are not only involved in transcriptional regulation, but also regulate the development of the thyroid gland (38, 39), it is possible that Id2 cooperates with these thyroid transcription factors in certain aspects of thyroid development.
Previous studies revealed that Id2 binds to the Rb protein and abolishes its growth-suppressing activity (20-22). However, we found that Id2 overexpression did not alter the phosphorylation status of Rb in response to TSH treatment (data not shown). The role of Id2 in TSH-mediated cell cycle regulation and thyrocyte proliferation should, therefore, be evaluated in future studies.
In summary, we have shown that Id2, one of the Id family proteins, is a major target for regulation by TSH. However, TSH-mediated Id2 expression did not alter the effects of TTF-1 and Pax-8 on the regulation of Tg gene expression.






 XML Download
XML Download