

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Regulation of the Executive Cells of Bone Remodeling by Osteocytes - the Sclerostin Paradigm
Osteocytes are ideally positioned to be the means by which bone adapts in response to mechanical stimuli. Osteoblasts and osteoclasts that are present on bone only transiently, in low number, and in variable locations. Osteocytes, on the other hand, constitute more than 90 percent of cells in bone and are strategically distributed throughout the entire bone volume. In addition, osteocytes form a syncytium among themselves and with cells on the bone surface via cytoplasmic processes that radiate from their bodies and travel along canaliculi excavated in the mineralized matrix. This network is perfectly suited to sense and respond to both mechanical and systemic stimuli by generating signals that affect osteoblasts, osteoclasts, and their progenitors in the bone marrow. In spite of significant progress in our knowledge about osteocytes in recent years, the mechanisms by which these cells control the function of osteoblasts and osteoclasts are just starting to emerge. Sclerostin is the first, undisputable mediator of the communication between osteocytes and the executive cells of bone remodeling. Osteocytes but not other cells in bone express sclerostin - the product of the Sost gene that antagonizes the action of Wnts and BMPs[4,5]. Evidence from human diseases and experimental animals indicates that sclerostin acts in a paracrine fashion to inhibit bone formation[4,6,7]. Recently, it was shown that sclerostin expression is potently inhibited by two recognized stimuli that increase osteoblast number: parathyroid hormone and mechanical loading[8~10], thereby representing a novel mechanism of regulation of bone formation mediated by osteocytes.
Osteocyte Apoptosis: Regulation and Consequences
That osteocytes perceive changes in the level of both physical stimuli as well as circulating factors is evidenced by studies on the regulation of their life span. Osteocytes are long-lived cells. However, like osteoblasts and osteoclasts, they die by apoptosis; and decreased osteocyte viability accompanies the bone fragility syndrome that characterizes glucocorticoid excess and estrogen withdrawal[11~13]. Conversely, preservation of osteocyte viability might explain at least part of the anti-fracture effects of bisphosphonates, which cannot be completely accounted for by changes in bone mineral density[14].
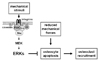
Osteocyte apoptosis is also regulated by mechanical forces. Thus, mechanical stimulation of osteocytes protects them from the pro-apoptotic action of glucocorticoids, etoposide and other death inducers[15,16]. Mechanistic studies indicate that the transduction of mechanical forces into intracellular signals is accomplished by molecular complexes assembled at caveolin-rich domains of the plasma membrane and composed of integrins, cytoskeletal proteins and kinases including the focal adhesion kinase FAK and Src, resulting in activation of the ERK pathway and osteocyte survival[15]. In vivo mechanical stimulation also regulates osteocyte life span. Thus, increased prevalence of apoptotic osteocytes is found in unloaded bones[1] or in bones exposed to high levels of mechanical strain[2]. In both cases, increased apoptosis of osteocytes was observed before any evidence of increased osteoclast resorption. Moreover, apoptotic osteocytes in unloaded bones accumulated in areas that were subsequently removed by osteoclasts[1]. Taken together with the in vitro evidence, these findings had suggested that diminished mechanical forces eliminate signals that maintain viability, thereby leading to osteocyte apoptosis; and that dying osteocytes in turn become the beacons for osteoclast recruitment to the vicinity and the resulting increase in bone resorption (Fig. 1).
Recent work provides direct evidence that death of osteocytes is sufficient to recruit osteoclasts and to increase resorption. Tatsumi et al.[3] generated transgenic (TG) mice expressing the diphtheria toxin receptor (DTR) under the control of the dentin matrix protein 1 (DMP1) promoter that is only active in osteocytes. DTR is normally not expressed in murine cells; therefore, osteocytes are the only cells sensitive to the toxin in these TG animals. A single injection of DT resulted in rapid induction of apoptosis of 70~80% of osteocytes; and this was followed by increased osteoclasts and loss of bone. These findings demonstrate that osteocyte apoptosis is sufficient to trigger osteoclast recruitment and bone resorption. Taken together with the evidence that osteocyte apoptosis is inhibited by estrogens and bisphosphonates[12,14], these findings also raise the intriguing possibility that preservation of osteocyte viability contributes to the anti-remodeling properties of these agents. Future research is required to directly test this stimulating hypothesis.
Osteocytes: Primary Culprits for the Bone Loss Induced by Physical Inactivity
Mechanical loading is critical for the maintenance of bone mass; and skeletal unloading as with reduced physical activity with old age, immobilization of bed rest, and total or partial motor paralyses, cause bone loss leading to disuse osteoporosis[17]. Furthermore, the bone loss that ensues under microgravity conditions represents the most significant hindrance for long-term space flying[18]. The rapid decrease in osteocyte viability with unloading had suggested that osteocytes are the first responders to the change in mechanical forces[1]. The results of Tatsumi et al.[3] demonstrate that mice depleted from osteocytes are protected from the bone loss induced by tail suspension indicating that in the absence of osteocytes bones are unable to elicit the normal osteoclastogenic response. These findings confirm that osteocytes are the primary culprit of the negative bone balance that ensues with weightlessness.
In conclusion, the osteocyte ablation model revealed that osteocyte apoptosis is sufficient to initiate an osteoclastogenic response and that osteocytes are required for the skeletal adaptation to reduced mechanical forces. Whether living osteocytes continually produce molecules that restrain osteoclast recruitment or whether in the process of undergoing apoptosis osteocytes produce pro-osteoclastogenic signals remains to be determined. It is expected that intense investigations will take place in the near future attempting to identify the molecular mediators involved in the communication between osteocytes and osteoclasts.
 XML Download
XML Download