

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
High blood pressure is a well-known, important risk factor for the development of atherosclerosis and a strong predictor of cardiovascular morbidity and mortality. Therefore, hypertension is one of the most important preventable causes of death all over the world, in particular, in East Asia. Untreated hypertension is associated with the incidence of ischemic and hemorrhagic stroke, myocardial infarction, heart failure, chronic kidney disease, and large vessel disease. In addition, hypertension is more popular disease with a prevalence accounting for 30% to 55% of population in developed countries.1) Because of escalating obesity, metabolic syndrome, and population aging in developed and developing countries, the global burden of hypertension is gradually rising. The clinical management of hypertension is the most common interventions in primary care since it proved to be an effective therapy for the prevention of cardiovascular disease. However, despite intensive efforts to prevent and treat hypertension, only less than one third of patients whose hypertension is assumed to be well-controlled by standard medication is protected from the future stroke, myocardial infarction, or heart failure.2)
Hypertension is currently defined as average systolic blood pressure ≥ 140 mm Hg and/or diastolic blood pressure ≥ 90 mm Hg, for which the benefits of pharmacological treatment have been definitely established in randomized placebo-controlled trial.3) Recently, according to new National Institute for Health and Clinical Excellence guidelines for hypertension, hypertension is described as follows: clinical blood pressure is 140/90 mm Hg or higher and subsequent ambulatory blood pressure monitoring daytime average or home blood pressure monitoring average blood pressure is 135/85 mm Hg or higher. Namely, the monitoring home blood pressure or ambulatory blood pressure is needed to diagnose hypertension. When the level of less than 135 mm Hg of systolic blood pressure or less than 85 mm Hg of diastolic blood pressure by the monitoring home blood pressure, the subjects seem to be normotensive or nonhypertensive.4,5)
Blood pressure variability
Although it has been more than half a century since blood pressure was first routinely assessed in the doctor's office, there is variability in blood pressure, and this is one of the reasons why hypertension cannot yet be successfully prevented or controlled as mentioned above.1) Diagnosis and treatment of hypertension depends on correct measurement of "true" blood pressure with individuals. However, this simple procedure is likely to pose several pitfalls, and the accuracy of evaluations for high blood pressure is often not good. In previous clinical studies, measured blood pressure in the doctor's office has been the standard, with multiple measurements permitting calculation of an average for diagnosis and treatment. Previous epidemiological studies show that an average of office measurements proved to be an independent risk factor for high cardiovascular morbidity and mortality. Currently, however, data on various aspects of blood pressure, observed in various settings, have been collected for analysis. Home-blood pressure measurement is feasible to identify so-called "white-coat" hypertension, and is proved to correlate with target-organ damage better than blood-pressure values measured in the doctor's office.6) A number of studies using the evaluation of patterns of blood pressure variability over time have been also reported to evaluate the precise risk of the current and long-term complications.7) At present, we can detect a variety of blood pressures under various conditions as well as blood pressure variability over time (Table 1); eventually, we would like to know which blood pressure, or its variability, is the most important measure for predicting the development of atherosclerosis and the occurrence of cardiovascular events. Referring to one of them, Matsui et al.8) recently reported that the maximum home systolic blood pressure measured in the morning and evening for two weeks had the best predictive value for hypertensive target organ damages, such as cardiac hypertrophy, carotid intima-media thickness, and albuminemia, suggesting that target organ damages by hypertension might be the most strongly associated with transient blood pressure fluctuations.
Blood pressure variability is also dependent on habits and behaviors. The nicotine in cigarette smoke transiently raises blood pressure by 10 to 20 mm Hg with every cigarette, thereby elevating the average in habitual smokers.9) With regard to alcohol, the risk of hypertension is lower in moderate drinkers drinking one or two drinks a day, whereas blood pressure is increased in heavier drinkers than those. On the other hand, caffeine consumption causes only a small transient rise in blood pressure.10)
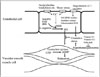
Blood pressure is always altered and significantly fluctuates from beat to beat in some cases, particularly in patients with arrhythmia. Essentially, blood pressure is physiologically controlled by several systems based on neural receptors such as baroreceptors, chemoreceptors, and central nervous system ischemic response, and is easily shifted to another level within seconds (Fig. 1).11) In addition, several hormonal and some minor systems modulate blood pressure within minutes. Finally, the kidney-fluid volume system affects blood pressure within hours or days. Guyton12) described how this latter factor contributes the most to controlling blood pressure after the arterial pressure suddenly becomes abnormal.
Mechanisms of essential hypertension
In young adults, essential hypertension often is associated with increased heart rate and cardiac output, released higher levels of norepinephrine, peripheral postganglionic sympathetic nerve firing, and alpha-adrenergic receptor-mediated vasoconstrictor tone in small resistance vessels. These mechanisms are involved in hypertension induced by reset baroreceptors, obesity, and sleep apnea.
The renal mechanisms are both culprit and victim in hypertension. There is a vicious cycle of progressive renal dysfunction and hypertension. It has been recognized that low birth weight increases the risk of developing adult salt-dependent hypertension. The reason is that low birth weight is suggested to be associated with deficits in nephron number in the infant kidney and with consequently increased risk of adulthood hypertension and renal dysfunction.13,14)
Activation of the renin-angiotensin-aldosterone system is another important mechanism contributing to hypertension as well as neural and renal mechanisms. Among several substances included in this system, angiotensin II possesses the effects of strong vasoconstriction, generation of reactive oxygen species, vascular inflammation, vascular and cardiac remodeling, and production of aldosterone, the principal mineralocorticoid.15)
Genetic contribution is involved in the mechanisms as mentioned above. There is a strong genetic contribution to hypertension, in which persons < 50 years of age with hypertension are 4 times more likely to have a first-degree relative with essential hypertension.16) Concordance of levels of blood pressure is reported to be much higher in families than in unrelated individuals, higher between monozygotic than dizygotic twins, and higher between biological than adoptive siblings living in the same household. It is known that as much as 70 percent of the familial aggregation of blood pressure is attributed to shared genes rather than shared environment.17)
Endothelial dysfunction and hypertension
1. Endothelial function
Endothelial dysfunction is thought to precede the clinical manifestations of atherosclerosis and is intimately associated with the development of atherosclerosis. Since nitric oxide (NO) was found as endothelium-derived relaxant factor at 1980 by Furchgott and Zawadzki,18) it has become evident that the endothelium is an active paracrine, endocrine, and autocrine organs that regulate vascular tone affecting blood pressure and structure and maintain vascular homeostasis. Moreover, the endothelium controls coagulation and inflammatory responses, and as a result, prevents the progression of atherosclerotic lesion involving pathological inflammatory process and consequently causing an increase in stiffness of vessels.19) These functions include impaired vasomotor disturbance, abnormal coagulation, and increased vascular proliferation (Table 2).
Endothelial dysfunction is defined as the endothelium characterized by vasomotor disturbances, abnormal coagulation fibrilolysis, altered local immune feature, and increased vascular proliferation, which plays an important roles in the pathogenesis of peripheral and coronary atherosclerosis. In particular, one of assessments determining endothelial dysfunction is physiologically estimated as an alteration of vasomotor activity. Namely, the disordered function in the based condition and inadequate response of the endothelium to mechanical and pharmacologic stimuli has been implicated as an early stage in the development of atherosclerosis20-23) and hypertension.24,25) The bioavailability of vasodilators is reflected not only by vasodilation effect via the relaxants such as NO and prostacyclin, but also by endothelium-derived contracting factors including endothelins and thrombosis A2. These imbalances lead to an impairment of endothelium-dependent vasodilation.
NO plays an important role in the regulation of vascular tone by production from endothelial cell and causing the relaxation of the vascular smooth muscle cells in the physiologic state (Fig. 2), and the impairment of endothelium-dependent vasodilation represents the functional characteristic of endothelial dysfunction and is thought to be chiefly attributed to decreased endothelial NO bioavailability. Contribution of NO to the endothelial dysfunction in atherosclerotic vessels involves not only impaired vasodilation but also smooth muscle cell proliferation and platelet and monocyte activities. Continuous inflammatory responses result in blanting vasomotor function19) and promoting atherosclerotic process.26) Therefore, reduced secretion of NO from the endothelium is deeply related with the development of atherosclerosis. In fact, we demonstrated that long-term inhibition of NO production by an NO synthase (NOS) inhibitor NG-nitro-L-arginine methylester (L-NAME) in drinking water promoted intimal atherosclerotic lesions in rabbit aortas.27) Although the plasma concentration of total nitrate/nitrite (NOx), the last product of NO, is thought to be a marker endothelial function, lack of associations between arterial wall endothelial NOS (eNOS or NOS3) levels and corresponding NOx levels suggests that eNOS may not be the only major enzymatic source for arterial wall NO productions.28)
2. Contribution of genomics to understanding essential hypertension and endothelial dysfunction
To date, huge efforts have been performed to find candidate-gene and genome-wide association for high blood pressure traits in general population samples and for case-control samples, and identified 47 distinct genetic variants at 40 foci strongly associated with blood pressure and hypertension.29-31) However, these variants explain only a few percent of the heritability for hypertension. Previous studies regarding genetic association with hypertension indicate the intimate link between an increasing effect on blood pressure and abnormal endothelial function. Recently, two studies have reported to identify blood pressure loci using a gene-centric array. They identified a new hypertension susceptibility locus of the single nucleotide polymorphism (SNP) (rs3918226) in the promoter region of the eNOS gene.32,33) However, Luizon et al.34) described this SNP does not affect nitrite formation in healthy subjects, suggesting that rs3918226 promotes hypertension by many mechanisms to be determined.34)
The assessment of flow-mediated dilation (FMD) has been proposed to represent a functional bioassay for endothelium-derived NO bioavailability in humans, and it has been recognized that endothelial dysfunction is an early event in atherosclerosis35) and hypertension.36,37) So far, extensive epidemiological evidence has consistently indicated that the endothelial dysfunction of the coronary artery plays a pivotal role in the development of atherosclerosis and cardiovascular disease.21) Similarly, it is possible that the assessment of endothelial function in the brachial artery may predict long-term cardiovascular risk regardless of the method used for the assessment.38-41)
In the previous studies, we demonstrated the impact of eNOS genotype on FMD in the brachial artery in the young, healthy men.42,43) It is well-known that eNOS has 3 common variants: -786T > C (rs2070744), 894G > T (Glu298Asp) (rs1799983), and 4b4a genotypes. Among three variants, two polymorphisms are characterized by a single nucleotide polymorphism: the 894G > T genotype is a missense mutation in the exon 7, and the -786T > C genotype is another one in the promoter region. The 4b4a genotype is a polymorphism divided by a number of tandem repeats in the intron. The -786T > C genotype is in strong linkage with the 4b4a genotype in our subjects (not reported) and even in other studies,44,45) whereas the 894G/T genotype is completely distributed independent of the -786T > C genotype in Japanese subjects. We demonstrated that two polymorphisms, the 894G/T and the -786T > C, are associated with variation in FMD in apparently healthy young men, who have not been less affected by the status of blood pressures, hypercholesterolemia, and hypertriglyceridemia than the older. Fig. 3 shows that the dominant effect of the 894T allele reduced FMD by 22% compared with wild-type haplotype as much as the dominant effect of the -786C allele (a 20% reduction compared with wild-type haplotype). The combination effect of the 894T allele and the -786C allele further reduced FMD by 48% compared with wild-type haplotype. In contrast, our study is disagreement with others who failed to find any significant association between eNOS gene polymorphism and endothelial function.46) Using a genome-wide analysis, Vasan et al.47) reported no association in FMD in the brachial artery among the polymorphisms of -786T > C, intron 4b4a, or 894G > T genotypes. However, the subjects recruited for their studies were quite older than ours.
In terms of the mechanism related to the difference in endothelial function, there have been several reports regarding each eNOS gene polymorphism. eNOS with asparate substituted for glutamate at position 298 in the 894G > T genotype was cleaved intracellularly in human endothelial cells and in human hearts, resulting in the generation of 100 and 35 kDa fragments.48) As a result, eNOS activity in the subjects with the 894T allele may be decreased, leading to decreasing endothelium-dependent vascular dilation, NO production, and susceptibility to cellular stress.49) Furthermore, this polymorphism is suggested to modulate eNOS activity by controlling its intracellular distribution.48) On the other hand, the mutant allele of -786T > C polymorphism is reported to affect the gene promoter activity, resulting in deficiency of NO production, which leads to increased vessel reactivity as observed in subjects with coronary vasospastic angina.50) In addition to endothelial dysfunction induced by both eNOS polymorphisms, the eNOS -786T > C polymorphism may be associated with plasma adiponectin levels and systolic blood pressure, whereas the 894G > T polymorphism may be associated with atherogenic lipid levels and insulin levels (Fig. 4).43)
The polymorphism of 5-10-methylenetetrahydrofolate reductase (MTHFR) gene is also a candidate of loci for phenotype of hypertension,32,51) and several polymorphisms involved in MTHFR gene are identified. An inherited thermolabile variant of MTHFR in the position 677 (MTHFR 677C > T) (rs1801133), alanine to valine substitution, reduces the specific activity of MTHFR, thereby modestly raising blood homocysteine levels.52,53) Furthermore, the 677T allele homozygous mutation lowers the blood folate levels probably due to a decrease in tetrahydrofolate synthesis by reduced remethylation pathway, as shown previously.53) Plasma folate levels are independently associated either with high density lipoprotein cholesterol levels or with FMD in the brachial artery.53) Both high homocysteine and low folate levels limit the bioavailability of NO,54,55) and increase oxidative stress56) (Fig. 2). These properties with the MTHFR 677T allele homozygous mutation are suggested to facilitate the proliferation of vascular smooth muscle cells resulting in atherosclerosis, and alter the elastic properties of the vascular wall resulting in hypertension. We also reported an adverse effect of the 677T allele on endothelial function assessed by FMD when the effect of the eNOS polymorphism is eliminated (Fig. 5).42)
The above two genes are involved in eight independent genetic variants associated with high blood pressure, which has been recently reported,30) and it is not surprising that some of the others might be related to reflecting impairment of endothelial function.
3. Experimental evidence of the association between endothelial dysfunction and hypertension
Acute inhibition of NO synthase induces peripheral vascular contraction, resulting in an increase in blood pressure.57) When we demonstrated that long-term inhibition of NO production with L-NAME promotes atherosclerosis in rabbits,25) we did not pay any attention to a change in blood pressure. However, it was already reported that long-term inhibition of NOS produces a sustained hypertension in rats58,59) and dogs,60) and this effect is able to be used as a new model of arterial hypertension. Rats treated by L-NAME showed an increase in systolic pressure from 120 mm Hg to 165 mm Hg, in part, via cyclooxgenase 2 (COX2) expression in kidney and aorta in addition to NO deficiency because hypertension was partly prevented by COX2 inhibitor.61)
4. Dietary supplementation for the purpose to produce nitric oxide and abolish endothelial dysfunction and high blood pressure
L-Arginine, a semi-essential amino acid, is the natural substrate for NOS and NO is generated from L-arginine and molecular oxygen in a reaction catalysed by NOS. It is speculated that supplemental oral L-arginine may enhance NO generation. Although it is conceived that physiological concentrations of L-arginine in healthy individuals are enough, there is evidence that L-arginine supplementation indicates NO-mediated effects in the endothelium in humans.62-64) The meta-analysis using randomized, double-blind, and controlled trials of oral L-arginine supplementation provide significant lowering systolic and diastolic blood pressures.65) This phenomenon is known as the "L-arginine paradox."66) In pathological conditions such as hypertension, diabetes mellitus, hypercholesterolemia, and kidney failure, plasma levels of asymmetric dimethylarginine (ADMA), an endogenous NO inhibitor, are increased.67) Such produced ADMA may be one possible explanation for endothelial dysfunction and decreased NO synthesis. Recently, acute L-arginine supplementation is not effective for an increase in NO production in healthy subjects.68) Therefore, it appears that only pathological conditions with poor NOS activity are likely to have benefit from L-arginine.
Nitrate and nitrite are found in considerable amounts in our diet, of which leafy green vegetables such as spinach and lettuce are particular rich in nitrate. Nitrate is the stable oxidation product of NO metabolism, and dietary nitrate is converted to nitrite via bioconversion in the entero-salivary circulation, and enters the systemic circulation. Nitrite can be further reduced to NO and other bioactive nitrogen oxides in blood and tissues.69,70) In animal model of unilateral nephrectomy and high-salt diet hypertension, dietary nitrate attenuated hypertension by restoring bioactive nitrogen oxides in the tissues and reducing oxidative stress markers like ADMA in plasma and urine.71) Not only L-arginine, but also nitrate is possible to be a candidate as a tool of dietary supplementation to produce NO for treatment for hypertension. Ingestion of dietary nitrate is reported to elevate plasma nitrite and cyclic guanosine monophosphate, a sensitive indicator of NO bioactivity.72) As a result, blood pressure was decreased in healthy volunteers.73,74)
5. Effect of antihypertensive agents on endothelial dysfunction
From the view of the effects on endothelial function, angiotensin converting enzyme inhibitors are superior to calcium channel blockers and beta-blockers in antihypertensive agents. Namely, according to a meta-analysis of studies using the assessment of FMD in the brachial artery, angiotensin converting enzyme inhibitors significantly improved endothelial dysfunction, and the beneficial effect on the endothelium is significantly more than that of calcium channel blockers or beta-blockers, but is as much as that of angiotensin receptor blockers.75) With regard to the effect of diuretics on FMD, combined treatment with perindopril and inadapamide is reported to be superior to atenolol.76)
6. NO production and vasodilation by the phosphatidylinositol 3-kinase
Activation of the phosphatidylinositol 3-kinase (PI3-kinase) is central in insulin-mediated glucose uptake and enhances eNOS phosphorylation and NO production.77,78) In metabolic abnormalities observed in subjects with type 2 diabetes mellitus and hypertension associated with obesity, activation of PI3-kinase by insulin is seriously impaired.79,80) This may be a reason why hypertension is developed in the insulin resistant states.
Under condition of endothelin-dependent hypertension, antidiabetic thiazolidinediones, like rosiglitazone or pioglitazone, prevent the vascular increase in superoxide anion production and progression of hypertension81) through activation of PI3-kinase.82) In humans, thiazolidinediones improve endothelial function in patients with type 2 diabetes mellitus or metabolic syndrome, as shown by increased flow-mediated, endothelium-dependent vasodilation.83,84) Thiazolinediones have also effect to partially prevent progression of hypertension in hypertensive animal models.81,85-87)
7. The ethnic difference in endothelial function
Epidemiological surveys demonstrate that the risk of development of hypertension is greater in black people compared to white people.88) Mechanistically, these ethnic differences have been attributable to an interplay of environmental and biological factors.89,90) Of these mechanisms, it is reported that the most important distinction of resistance vessel properties seems to be the reduced vasodilation and reduced NO bioavailability in the resistant vessels.91)
Relation between blood pressure variability and endothelial dysfunction
Asymptomatic, obese adults with circadian blood pressure abnormalities such as elevated blood pressure and pulse pressure show attenuated FMD assessed in a fasting state between 8 and 10 a.m. in the brachial artery.92) Recently, even increased blood pressure variations estimated by visit-to-visit systolic blood pressure variability, 24-hour systolic blood pressure variability, and 24-hour diastolic blood pressure variability was associated with reduced endothelial function as well as reduced vascular smooth muscle function.93) It is also reported that the maximum home systolic blood pressure measured in the morning and evening for 14 consecutive days had the best predictive value for hypertensive target organ damages.8) These results indicate that blood pressure variability during a day or a week is an important, new risk factor for cardiovascular disease and suggest that restoring impaired endothelial function is a target to prevent blood pressure variation and future cardiovascular events. The pathophysiological basis of the relationship between blood pressure variability and endothelial dysfunction remains to be elucidated.
Conclusions
There are several aspects of blood pressure. How to best assess blood pressure average and variability is still a matter of the ongoing debate on the clinical value of blood pressure.94) We must pay more careful attention focused on hypertension with blood pressure fluctuation. In order to predict the future atherosclerosis and cardiovascular events in subjects with hypertension, the adequate assessment of endothelial function is one of the most reliable markers.






 XML Download
XML Download