

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Primary sclerosing cholangitis (PSC) is a rare progressive disease of the liver characterized by cholestasis and bile duct fibrosis, leading to decreased quality of life, cirrhosis, and the need for liver transplantation within an average of 12 years [1]. The disease occurs in children and adults, and is closely linked with inflammatory bowel disease (IBD), most often colitis, especially ulcerative colitis (UC) [1]. Small-duct PSC has an overall better long-term prognosis than large-duct PSC [2]. While the pathogenesis of PSC remains obscure, a leading theory is that an abnormal gut microbiome activates innate immunity within the liver, resulting in bile duct-targeted inflammation and biliary fibrosis [1]. Presently, there are no effective therapies known to delay the progression of PSC. In this case, a single antibiotic, oral vancomycin, led to normalization of the patient's liver enzymes.
CASE REPORT
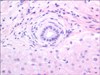
The patient first presented at age 13 with bloody diarrhea after taking doxycycline daily for acne treatment 4 months prior to initiation of symptoms. She was diagnosed with blastocystis hominis and treated with ciprofloxacin and metronidazole (for 10 days), and later with nitazoxanide (for 4 days). Her symptoms improved while on the ciprofloxacin/metronidazole, but returned when the treatment was completed. She was admitted to the hospital 3 months later with erythema nodosum which resolved without treatment. She discontinued the doxycycline treatment just prior to her hospitalization. Her labs showed slight elevations in alanine aminotransferase (ALT), alkaline phosphatase (ALP), and perinuclear anti-neutrophil cytoplasmic antibodies were positive at 113.9 EU/mL. The patient was diagnosed by colonoscopy with moderate chronic active ulcerative pancolitis and was started on mesalamine, which she did not tolerate and had worsening bloody diarrhea and abdominal pain. She was transitioned to 9 mg of budesonide and later also to 6,750 mg/day balsalazide, the latter which also caused worsening diarrhea. The patient elected to stop all medication and started herbal remedies for 6 months: VSL#3 probiotic, curcumin, and Nopalea (cactus juice; Trivita in Scottsdale, AZ, USA). The patient continued to experience diarrhea up to 4-5 times a day and azathioprine was initiated. Screening blood tests for azathioprine therapy in August 2012 revealed increased liver enzymes (Fig. 1). Quantitative immunoglobulin G (IgG) was normal and screening for hepatic autoimmune antibodies (anti-nuclear antibodies, anti- smooth muscle antibodies, and anti-liver/kidney microsomal antibodies) was negative. The IgG subclasses panel showed normal IgG4. A magnetic resonance cholangiopancreatography (MRCP) showed localized common hepatic duct prominence measuring 7 mm proximally at the porta hepatis of uncertain significance with no changes of PSC. A liver biopsy showed mild portal lymphocytic infiltrates with focal infiltration of bile duct epithelium. Several small and medium-size bile ducts were surrounded by concentric fibrosis with mild bile ductular proliferation. There was no significant fibrosis. Findings were consistent with small-duct PSC (Fig. 2).
With the diagnosis of PSC, the patient was started on ursodeoxycholic acid (UDCA) 300 mg 2× per day. The liver enzymes improved but did not normalize (Fig. 1). She developed intolerance to azathioprine: nausea, vomiting, and epigastric pain, which resolved with discontinuation of the medicine. Over the next two months, she continued to have diarrhea and malaise. She was started on 500 mg oral vancomycin 3× per day (35 mg/kg) for the management of PSC as the patient was reluctant to start infliximab. The diarrhea resolved within three days, and after several weeks stools became solid. UDCA was discontinued after starting oral vancomycin.
The patient gained weight, fatigue significantly improved and within 2 months, the patient began menarche. After 9 months of therapy, transaminases had not normalized, and the dose of vancomycin was increased to 750 mg 3× per day (40 mg/kg) leading to normalization of liver enzymes with only mild gamma-glutamyltransferase (GGT) elevation (Fig. 1). Patient's surveillance MRCPs in 2 and 3 years following the diagnosis showed a normal liver with resolution of the localized hepatic duct prominence and normal bile ducts. Surveillance colonoscopies in each subsequent year showed nearly normal, with only quiescent to mild chronic colitis on biopsy. Vancomycin dose was reduced to 1,000 mg 2× per day (35 mg/kg) and her liver function tests (LFTs) remained fully normalized. The patient has continued on this dose to date and it is well tolerated. She has had no infections. She has continued to have normal bowel movements and normal LFTs, other than transient elevations in the LFTs when she contracted acute infectious mononucleosis (Fig. 1).
DISCUSSION
PSC, a rare progressive liver disease characterized by cholestasis and bile duct fibrosis, has no accepted, effective therapy known to delay or arrest its progression. UC is an IBD of unclear etiology characterized by diffuse mucosal inflammation of the rectum and the colon. At diagnosis, pediatric UC associated with PSC is often more severe and extensive and many patients are affected with pancolitis. Pediatric UC can be very aggressive with a high frequency of acute severe exacerbations. Patients with UC are known to develop PSC in 4-5% of cases [3], therefore, patients with IBD should be screened for abnormal liver enzymes. Other causes of abnormal LFTs in patients with IBD may be autoimmune hepatitis, drug hepatotoxicity, disease flare, and malnutrition.
UDCA has been extensively utilized as a therapeutic option for the treatment of pediatric PSC. One uncontrolled pilot study showed that UDCA improved liver biochemistries, however there is no evidence that UDCA alters the progression of disease [45]. A multicenter study of adults with PSC using high-dose UDCA (28-30 mg/kg/day) over 5 years showed improvement in liver biochemistries, however the study had to be terminated because of an increased risk of death and liver transplantation in the UDCA group [6].
Vancomycin is a glycopeptide antibiotic with bactericidal activity against many gram-positive bacteria. Patients with UC and PSC who are treated with oral vancomycin have been shown to improve both UC symptoms and liver biochemistries [789], suggesting that oral vancomycin could be of therapeutic benefit in patients with PSC-UC. A study with 14 pediatric patients with PSC and concurrent active IBD treated with oral vancomycin showed significant improvement in GGT, ALT, and erythrocyte sedimentation rate values within 3 months of therapy [8]. These patients also measured higher levels of transforming growth factor-β, an anti-inflammatory protein, and regulatory T cells (inhibits autoimmunity) indicating a potential immunoregulatory mechanism of action of vancomycin [7]. Oral vancomycin treatment was also effective in the treatment of a pediatric patient with recurrent PSC after an orthotopic liver transplantation, suggesting a disease mechanism with some causes external to the liver—potentially from the gut bacteria [10].
In children and adolescents, ALP may be elevated due to accelerated growth and bone metabolism, therefore GGT is a followed as a marker of biliary disease. The patient had a diagnosis of small duct PSC and had no fibrosis at diagnosis, which generally has a more benign course [2]. An additional benefit associated with the oral vancomycin treatment was the resolution of the patient's UC symptoms and colonic mucosal healing. The oral vancomycin may be acting both as an antibiotic by altering the intestinal microbiome and as an immunomodulator. The microbiome has been shown to affect a spectrum of liver diseases as intestinal blood flows through the portal tract. Modulating the microbiome through oral vancomycin, which mostly affects gram positive bacteria, may have resulted in decreased PSC activity and normalization of GGT. Another mechanism is a direct immunomodulatory effect of oral vancomycin. Oral vancomycin is given orally with minimal systemic absorption, therefore the effect is likely via the intestines. Based upon this single case report and a case series, oral vancomycin may be a promising treatment for PSC that needs to be further studied in randomized trials.

 XML Download
XML Download