

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Colorectal cancer (CRC) is one of most prevalent malignancy in the world.1 The occurrence of CRC has increased steadily in recent decades, particularly in Eastern Europe, Latin America, and Asia.1 Most CRC occurs sporadically due to genetic mutations and epigenetic modifications of human genome.2 These genetic mutations and epigenetic modifications drive the progression from normal mucosa toward carcinoma by altering signaling pathways that regulate behaviors of cancer. Genetic and epigenetic alterations were originally established as independent mechanisms contributing to colorectal carcinogenesis. However, recent evidences indicate a crosstalk between these 2 mechanisms during colorectal carcinogenesis. Genetic mutations enable modification of several epigenetic controls while epigenetic modifications allow genomic instability and mutagenesis.3 Recently commercialized next-generation sequencing (NGS) have revealed unexpected genetic mutations associated with epigenetic alterations in various cancers. These mutations have the capability to modify cytosine methylation, histone modification, and nucleosome organization. In the meantime, epigenetic silencing of DNA mismatch repair (MMR) genes frequently contribute to genomic instability and lead to mutations of oncogene or tumor suppressor genes.4
GENOMIC INSTABILITY OF CRC
Genomic instability includes various genetic or genomic changes ranging from point mutations to chromosomal rearrangement.5 Cytogenetic studies have shown frequent genomic instability in CRC samples. Genomic instability is a definite characteristics of CRC carcinogenesis with 2 distinct pathways: chromosomal instability (CIN) and microsatellite instability (MSI). CIN has been found in approximately 85% of CRC while and the remaining 15% of CRC might have MSI.6
1. Adenoma-Carcinoma Sequence
Vogelstein proposed a multistep carcinogenesis of CRC in 1990 and demonstrated sequential genetic mutations that collaborated in the conversion from normal colonic epithelium to adenocarcinoma.7 This multi-step carcinogenesis comprises genetic mutations in several characteristic genes, such as loss-of-function in APC gene (5q) as an early event of multistep carcinogenesis at the stage of development of adenoma, gain-of-function of KRAS oncogene (12p12) for the progression to large adenoma, and loss-of-function of tumor suppressor genes TP53 (17p) and DCC (18q) for the transition from large adenoma to adenocarcinoma.7 Metastatic lesion can gain additional genetic alterations such as loss of heterozygosity (LOH) on 10q or gains of DNA sequences at 5p and 6p.8
2. Chromosomal Instability
Characteristics of CIN are aneuploidy (abnormal chromosome number) and LOH in cells.9 Although underlying mechanisms of CIN during colorectal carcinogenesis remain unknown, CIN might be caused by chromosomal segregation and defective response to DNA damage that can disturb tumor suppressor genes and activate oncogenes.59
Loss of 17q is commonly identified in CRCs. In situ chromosomal hybridization has identified human TP53 gene positioned on the chromosome 17 long arm (17q21–q22). TP53 is an important cell cycle checkpoint gene. It is known that TP53 inactivation causes uninhibited access in the cell cycle.9
TP53 mutation allows excessive cell proliferation, driving tumor progression and progressing adenoma into invasive carcinoma.79
TP53 mutation is more frequent in non-hypermutated CRCs compared to that in hypermutated CRC which has similar phenomenon in APC mutation.10
APC mutations result from flaws in chromosome segregation11 that activates WNT signaling as an initial critical step during colorectal carcinogenesis. In Cancer Genome Atlas (TCGA) study, WNT signaling is activated in most CRCs.10 Genetic disruption of APC gene that activates WNT signaling has been identified in approximately 80% of CRCs. APC induces proteasomal degradation of CTNNB1. However, mutant APC fails to degrade CTNNB1 which will accumulate in the cytoplasm, transfer into the nucleus, modulate transcriptional activity, and activate MYC and many other oncogenes. In addition to chromosome segregation, APC regulates cell adhesion, differentiation, migration, and apoptosis. Only 5% to 10% CRCs show genetic/epigenetic alterations on genes such as CTNNB1 involved in other WNT pathways.10
Loss and/or LOH of 18q is 1 common genetic alteration in CRCs. It might be because 18q includes SMAD2, SMAD4, and SMAD7 that are transcriptional mediators of transforming growth factor β (TGF-β) signaling.12 TGF-β signaling regulates cell growth, differentiation, and apoptosis, and promotes MYC activation.10 Besides these CINs, the gain of chromosomes 7, 8q, 13q, and 20q and the loss of chromosomes 1p, 4, 8p, and 22q have been reported.31314 CIN is associated with unfavorable outcome of CRC. It can be considered as a prognostic marker.15
3. Microsatellite Instability
Lynch syndrome is caused by genetic mutations in DNA MMR genes including MLH1, MSH2, MSH6, and PMS2. The loss of MMR protein results in genomic instability with numerous genetic alterations frequently found in non-encoding microsatellite regions.16 National Cancer Institute (NCI) recommends MSI status evaluation with a panel of 5 Bethesda markers (3 dinucleotide repeat markers: D2S123, D5S346, D17S250, and 2 mononucleotide repeat markers: BAT 26 and BAT 25) in CRCs.17 MSI status of CRCs can be classified into MSI-high (MSI-H, the presence of instability at least 2 Bethesda markers), MSI-low (MSI-L, the presence of instability in 1 Bethesda marker), or microsatellite stable (MSS; no marker presence). MSI-L and MSS CRCs can be categorized as 1 subtype because they have no significant difference in prognosis.18 Short repetitive sequences within coding regions of tumor suppressor genes can result in loss-of-fuction.19 Although there is mutual exclusivity between CIN and MSI, most CRCs are thought to arise from a similar adenoma-carcinoma sequence. In human MSI-H CRC and cancer cell lines with MSI-H, APC, KRAS, and TP53 have been found to be mutated.2021
MSI-H tumors have been found in about 15% of sporadic CRCs. They are associated with hypermutated subtype of CRCs (defined as cancers with mutation rates of >12/106 bases).10 Although MMR-defective status in 20% of MSI-H CRCs might be due to frameshift mutations or other multiple mutations of MMR gene, 80% of MSI-H CRCs achieve MMR-defective status by biallelic hypermethylation of MLH1 gene. Inactivation of MMR genes can induce MSI-H status with extensive methylation of the promoter region of CpG islands without inducing losses or gains in the chromosomal region (low copy number alteration). These MSI-H CRC have frequent BRAFV600E mutation but infrequent APC and TP53 mutations.
Recent exome sequencing of CRCs and endometrial cancers has revealed significant numbers of non-silent somatic mutations in RNF43 with high prevalence of MSI-H.22 RNF43 is known as an E3 ubiquitin-protein ligase that inhibits WNT signaling.23 A subsequent study has found that RNF43 mutation is accompanied by BRAF mutations in serrated pathway.24
MSI-H CRC tends to occur in females, the elderly, and the proximal colon. MSI-H CRC shows favorable response to 5-fluorouracil (FU).2526 MSI and/or MMR-deficiency can increase somatic mutations in tumor cells, leading to increased neoantigens expression. These changes are associated with abundant tumor-infiltrating lymphocytes and increased susceptibility to checkpoint inhibitors.27 Recently, U.S. Food and Drug Administration has approved pembrolizumab, a monoclonal anti-programmed cell death protein, for MSI-H and MMR-deficient cancers. The response rate to pembrolizumab in 149 patients with MMR-deficient or MSI-H cancers was 39.6% (complete response, 7.4% and partial response, 32.2%).28
4. KRAS Gene
RAS proto-oncogenes (HRAS, KRAS, and NRAS) regulate key cellular signaling pathways including phosphoinositide-3 kinase (PI3K) and mitogen-activated protein kinases (MAPK) pathways.29 Mutations in any RAS gene found in 20% to 25% of all human tumors, of which KRAS mutation accounts for about 85%. In CRC, KRAS mutation occurs in 30% to 50% of CRCs while NRAS mutations only occur in 2.5% to 4.5% of CRCs.30 Primary KRAS mutations will result in hyperplastic changes. However, when KRAS mutations are followed by APC mutation, adenoma progresses to cancer.31
KRAS and NRAS mutations are accepted as predictors of epidermal growth factor receptor (EGFR) inhibitors for treatment of CRC. Monoclonal antibodies to EGFR can induce receptor internalization followed by inhibition of tyrosine kinase and the downstream MAPK signaling. However, KRAS mutations can active guanosine triphosphate-bound protein which subsequently leads to “switch on” of downstream signals permanently. Therefore, CRC with mutant KRAS are resistant to anti-EGFR antibody such as cetuximab and panitumumab.32
Point mutations of KRAS in CRCs are commonly located in codons 12 (82%–87%) and 13 (13%–18%) but infrequently found in codon 61, 63, or 146.33
KRAS mutations might be associated with the phenotype of CRC.34 The association between KRAS mutations status and prognosis in patients with CRC has been controversial.30 Mutation in KRAS codon 12 or BRAF is enriched in proximal CRCs whereas wild-type KRAS/BRAF is increased in distal CRCs.35 KRAS codon 12 mutation including KRASG12C and KRASG12V could be associated with poor survival rate in advanced and recurrent CRCs.36
5. TP53 Mutations
Tumor protein p53 (also known as p53) encoded by TP53 gene is crucial in multicellular organisms as a main cell cycle checkpoint regulator. It prevents cancer formation and progression.9 Hence, TP53 is classified as a tumor suppressor gene. TP53 inactivation allows excessive cell proliferation that drives tumor progression. Traditional concept for multistep carcinogenesis is that TP53 is mainly associated with the evolution from adenoma to invasive cancer. This concept is advocated by frequent LOH or loss of 17q. TP53 mutation is more prevalent in non-hypermutated CRCs than that in hypermutated CRC.10 In addition to loss-of-function, gain-of-function of TP53-mutants also promotes tumor progression and invasion by tumor metabolic reprogramming.37
EPIGENOMIC INSTABILITY OF CRC
Epigenomic instability defined as aberrant response in gene expression regulation to environmental variabilities.38 CpG islands methylation in the promoter region of specific gene might alter chromatin conformational structure and DNA accessibility of the transcription apparatus, thereby regulating gene expression.39 Hypermethylation of CpG islands usually prevents expression of a certain gene, including tumor suppressor gene.3
1. CpG Island Methylator Phenotype
Epigenomic instability characterized by multiple CpG islands hypermethylation is referred to as CpG island methylator phenotype (CIMP). CIMP-positive tumors show hypermethylation in promoter regions of tumor suppressor genes that can lead to loss-of-function of genes. CIMP-positive CRCs develop via a serrated pathway.40 However, the prognostic role of CIMP in CRC patients remains contradictory between studies. Such discrepancy might be due to differences in definitions of CIMP.41 Specific CIMP definitions have not been established yet.42 Most studies have reported more unfavorable outcome in patients with CIMP-positive/CIMP-high CRC than those with CIMP-negative/CIMP-low CRC.41 Despite of no association between CIMP status and CRC survival in recent population-based study,42 combinations of CIMP with MSI status or BRAF mutation are associated with CRC survival.41 Most CIMP-positive CRCs reveal MSI-H tumors. Meanwhile, CIMP-positive CRCs with MSS have higher frequency of BRAFV600E mutation. They tend to develop in the proximal colon in later age of females and show poor tumor differentiation.
2. BRAF Mutation
Approximately 8% of CRCs have a point mutation in BRAF such as BRAFV600E and BRAFD594G that is mutually exclusive with KRAS mutations. Patients with BRAFV600E mutation are prevalent with proximal CRC and unfavorable outcome. BRAF mutations such as KRAS and PIK3CA are involved in MAPK and PIK3 pathways in colorectal carcinogenesis.43
BRAF mutations also show resistance to anti-EGFR antibody treatment.44
CLASSIFICATION BY MOLECULAR SUBTYPE
Based on several distinct molecular entities that have been defined, biologically distinct subgroups with their own clinical course have been proposed.45 Due to recent rapid evolution of high-throughput sequencing technologies such as genome-wide association study, whole exome sequencing, whole genome sequencing, and RNA sequencing, we can generate large-scale sequencing data for genetic and epigenetic alterations of CRCs. High-throughput sequencing data sets can be integrated to enhance information extraction using sophisticated bioinformatics software.
1. Classification Based on Clinical, Morphological, and Molecular Features
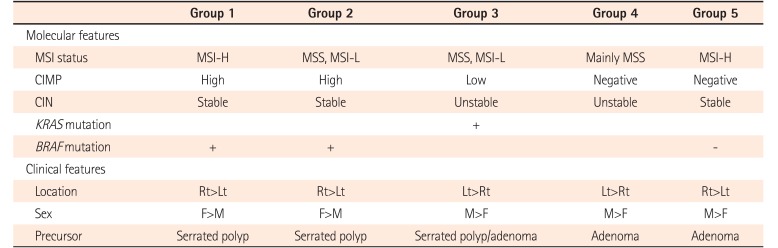
Primarily based on status of CIN and DNA methylation, the following 5 molecular subtypes of CRC have been suggested:46 (1) group 1, MSI-H, CIMP-high, methylation of MLH1, chromosomally stable, and BRAF mutation (sporadic MSI-H CRC, 12%); (2) group 2, MSS/MSI-L, CIMP-high, partial methylation of MLH1, chromosomally stable, and BRAF mutation (8%); (3) group 3, MSS/MSI-L, CIMP-low, CIN, KRAS mutation, and MGMT methylation (20%); (4) group 4, mainly MSS, CIMP-negative, and CIN (sporadic and Familial adenomatous polyposis/MUTYH polyposis-associated CRC, 57%);47 and (5) group 5, MSI-H, CIMP-negative, chromosomally stable, and BRAF mutation-negative (Lynch syndrome and familial MSI-H CRC, 3%) (Table 1).
These molecular subtypes are defined based on precancerous lesion and progression of CRC with discrete molecular features. Groups 1 and 2 CRCs may originate from serrated polyps whereas groups 4 and 5 CRCs may arise from adenomatous polyp through adenoma–carcinoma sequence. Group 3 CRCs may develop from adenomatous or serrated polyp. Groups 1 and 4 are considered as having few overlaps of molecular features with each other whereas groups 2, 3 and 5 share molecular features with groups 1 and 4 in different ways.
2. The Cancer Genome Atlas Classification
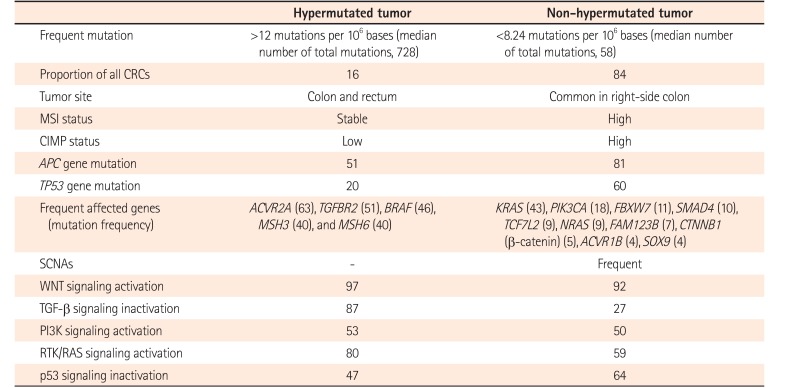
In 2012, TCGA performed genome-wide analysis for 276 CRCs, including exome sequence, DNA copy number variation (CNV), CpG island methylation, mRNA expression, and microRNA expression (Table 2).10 Exome sequencing has been performed for 224 tumor/normal pairs and 97 subset samples have undergone low-depth-of-coverage whole-genome sequencing. CRCs with >12 mutations/106 bases are defined as hypermutated CRCs (16%) whereas CRCs with <8.24 mutations/106 bases are referred to as non-hypermutated CRCs (84%).
In hypermutated CRCs, three-fourths of them have the expected MSI-H usually with MLH1 hypermethylation and CIMP-positive whereas one-fourth of them have somatic MMR mutations and POLE mutations with >40 mutations/106 bases (ultramutator phenotype). Hypermutated CRCs show frequent mutation in ACVR2A, APC, TGFBR2, BRAF, MSH3, and MSH6 genes. All non-hypermutated CRCs are characterized by MSS and frequent somatic copy number alterations (SCNAs), suggesting that these tumors are associated with chromosomal and sub-chromosomal alterations. Genomic alteration and somatic mutations in non-hypermutated CRC are enriched in APC, TP53, KRAS, PIK3CA, FBXW7, SMAD4, TCF7L2, NRAS, FAM123B, CTNNB1 (β-catenin), ACVR1B, and SOX9 genes. TP53 and APC genes in hypermutated CRCs are mutated less frequently compared to those in non-hypermutated CRCs (20% vs. 60% and 51% vs. 81%, respectively).
WNT signaling is activated in most types of CRCs (94% in non-hypermutated CRCs and 97% in hypermutated CRCs) by either APC inactivation or CTNNB1 activation. TGF-β pathway is inactivated in most hypermutated CRCs (87%) compared to that in non-hypermutated CRCs (27%). WNT signaling activation and TGF-β signaling inactivation will result in MYC activation.
There are no significant biological differences in CNV, expression profile, CpG methylation, or miRNA changes between colon and rectum of non-hypermutated CRCs. However, right-side colon cancers are more likely to be hypermethylated with high frequency of mutation compared to left-side cancers. In addition, TCGA study has suggested CRCs therapeutic approaches, focusing on WNT signaling4849 and MAPK/PI3K signaling.10
3. Molecular Classification Based on Gene Expression Profiles and Clinical Response to EGFR Inhibitor, Cetuximab
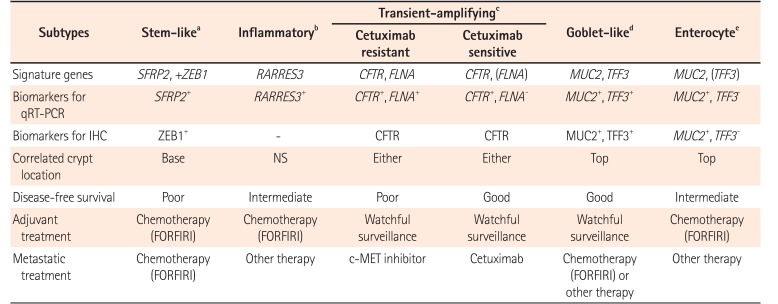
Based on gene expression profiles and therapeutic response to cetuximab in 80 patients, the following 6 clinically relevant CRC subtypes have been identified: stem-like, inflammatory, cetuximab resistant transient-amplifying (CR-TA), cetuximab sensitive transient-amplifying (CS-TA), goblet-like, and enterocyte subtypes. These subtypes share similarities with different cell types in colonic crypt, indicating that each subtype is associated with stemness and Wnt signaling. These subtypes can be classified by specific gene profile (Table 3).50 Goblet-like, CS-TA, and CR-TA subtypes have favorable outcome after surgical resection, suggesting tolerable chemotherapy. CR-TA subtype does not respond to cetuximab. It can be identified by filamin A expression. Metastatic CRC with CR-TA subtype may respond to c-MET receptor tyrosine kinase inhibitors.50
4. Consensus Molecular Subtypes of CRC Classification
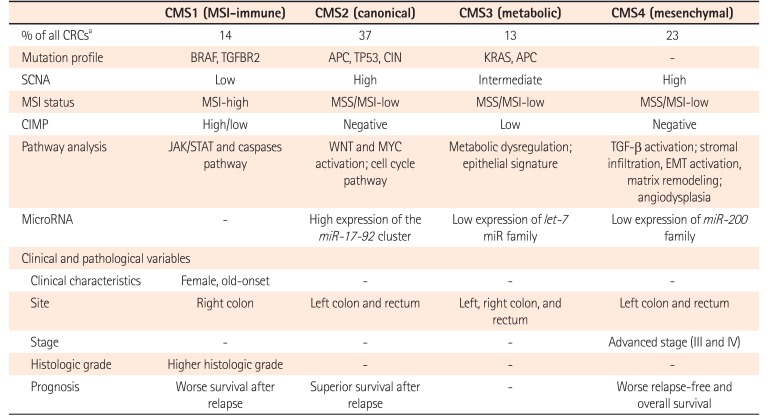
Although several studies have used gene expression profile for CRCs classifications, results are inconsistent. They fail to provide useful single classification.50515253 To overcome this limitation, an international Colorectal Cancer Subtyping Consortium has shared and analyzed a large-scale data for 4,151 patients with CRC. After applying unsupervised clustering techniques in analytics, they are categorized into 4 consensus molecular subtypes (CMS) (Table 4).54 CMS1 is characterized by immune activation, MSI-H, mutation in BRAF and TGFBR2, SCNA-low, CIMP-positive, and activation in Janus kinase/signal transducers and activators of transcription (JAK/STAT) and caspases pathway (14%). CMS1 CRC tend to occur in the elderly, female, and proximal colon with worse survival after relapse. CMS2 is characterized by canonical feature with mutation in APC and TP53, CIN, SCNA-high, MSS/MSI-L, CIMP-negative, and marked WNT and MYC pathway activation (37%). CMS2 CRC appear to occur in the left-side colon and rectum with superior survival after a relapse. CMS3 is characterized by metabolic features with mutation in KRAS and APC, MSS/MSI-L, SCNA-intermediate, CIMP-low, and exhibiting an epithelial signature and metabolic dysregulation (13%). CMS4 is characterized by mesenchymal feature with SCNA-high, MSS/MSI-L, CIMP-negative, and activation of TGF-β signaling. CMS4 CRCs seem to be diagnosed with advanced stages, showing poorer overall survival (23%). Although CMS classification cannot suggest a therapeutic stratification, extensive datasets can facilitate the understanding of complexity in molecular features of CRC.
5. CRC Subtypes Based on Combinations of Tumor Markers
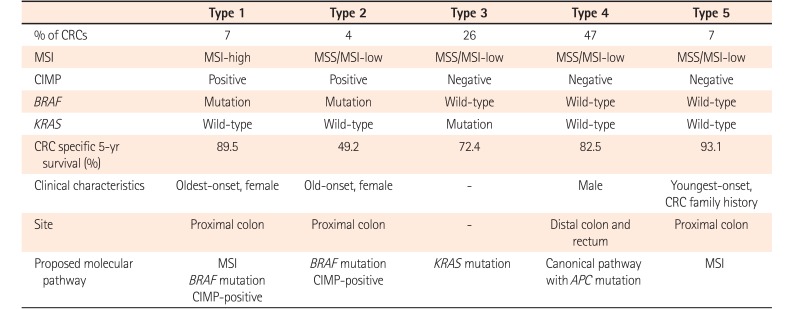
Development of CRCs involves distinct pathways combined with different genetic and epigenetic alterations. Based on specific combinations of MSI, CIMP, and mutations in BRAF and KRAS, traditional, alternate, and serrated pathways have been proposed in colorectal carcinogenesis.4655 CRCs arising from “traditional” adenoma-carcinoma sequence are characterized by MSS/MSI-L and CIMP-negative without BRAF mutation. CRCs originated from a “serrated” pathway are characterized by CIMP-positive and frequent BRAF mutation. CRCs with additional “alternate” pathway is characterized by MSS/MSI-low, CIMP-low, and KRAS-mutation.5556 Although MSI, CIMP, BRAF-, or KRAS-mutations have been studied extensively, the significance of combinations of these markers has been unclear.56 A total of 563 incident CRCs obtained from a population-based Women's Health Study can be assigned to 5 subtypes with distinct clinicopathological features (Table 5).57 CRCs with type 1 or 5 (MSI-H) showed favorable survival whereas those with type 2 (MSS/MSI-L, CIMP-positive, BRAF-mutation, KRAS wild-type) had unfavorable outcome. Type 2 CRCs are clinicopathologically characterized by the proximal colon location and females with late age onset. Type 5 CRCs are referred to as MSS/MSI-L, CIMP-negative, wild-type BRAF and KRAS CRCs. They have the lowest mortality rates. Type 5 CRCs show proximal colonic location and the youngest onset. Type 4 CRCs, the most prevalent subtype (47%), are characterized by MSS, CIMP-negative, BRAF and KRAS wild-type. Type 5 CRCs are clinicopathologically associated with canonical WNT pathway with APC mutations. They appear to have frequent occurrence in men and in the distal colon and rectum.
LIMITATIONS OF MOLECULAR CLASSIFICATION IN CLINICAL IMPLEMENTATION
Sometimes, it is difficult to define a combination of genetic markers representing a specific subtype of CRC. High throughput techniques provide comprehensive molecular characteristics and allow reclassification of CRC. However, high-throughput data from NGS also show heterogeneous molecular features even for the same CRC sample due to tumor heterogeneity. Tumor bulk consists of diverse cell types with distinct molecular signatures. Intratumoural heterogeneity might be due to genetic variation, stochastic processes, the microenvironment, and cell/tissue plasticity.58 Emerging evidences indicate that tumor heterogeneity provides fuel for resistance to current genetic/epigenetic alteration-guided strategies for anti-cancer therapy.59
Although the number of patients eligible for genome-target therapy has increased over time, medications used for genome-target therapy have only helped a small number of patients with advanced cancer. A cross-sectional study using publicly available data in United States suggested that fewer than 16% of patients were eligible for genome-target therapy while fewer than 7% of patients would benefit from genome-targeted cancer drugs in 2018.60 Current classifications by molecular subtype in CRC are able to improve CRC outcome only in a small portion of patients. This might be due to potentially different classification marker sets or methods, insufficient validation studies, and few evidences of the cost-effectiveness from this still high-cost technique. In addition, evolving technologies have generated vast amounts of molecular biological information which may dilute the meaning of current molecular classifications.
CONCLUSIONS
Recent high-throughput analyses regarding comprehensive molecular characterizations of CRCs have enlarged our understanding of their genomic and epigenomic landscapes which have enabled CRCs to be reclassified into biologically and clinically meaningful subtypes.21 In CRCs, genetic and epigenetic events are not indifferent phenomenon. They cooperate for CRC carcinogenesis, although methylation events are more common than point mutations. Integration of genetic and epigenetic alteration in CRC might embody the potential tool for proper diagnostic, prognostic, and therapeutic approaches. Moreover, the identification of key molecular features or pathways specific to a certain CRC subtype may represent potential therapeutic targets, enabling implementation of tailored therapies with better patient management.61
However, heterogeneity can provide seeds for resistance to genome-target therapy. Future cancer therapy should focus on the eradication of heterogeneity. Molecular characteristic exploration of clonal dynamics from tissue samples obtained from resistant site for conventional treatment have most promising guide to the development of treatment strategies that address tumor heterogeneity.58 However, tissue samplings at regular intervals and from multiple sites are major limitations. Theoretically, noninvasive liquid biopsy sampling enables frequent and comprehensive surveillance. NGS produces high-throughput information and demonstrates excellent testing performance using low-input DNA. Integration of NGS with liquid biopsy can maximize overall advantages. NGS-based liquid biopsy might enable minimally invasive and comprehensive genomic profiling of CRC that overwhelms spatial heterogeneity arising from tissue biopsy and limitations in genomic information obtained from candidate gene characterization.62
There is no doubt that an approach based on high throughput molecular information is beneficial for tailored therapies and individualized management in response to the complexity of CRC and the seemingly endless arc of evolution. Further research and validation are urgently needed to adopt these molecular classification systems into clinical practice.
 XML Download
XML Download