

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Adams-Oliver syndrome (AOS) is a rare congenital anomaly complex characterized by aplasia cutis congenita in the vertex area, terminal transverse defects of the limbs, and cutis marmorata telangiectatica congenital (CMTC). As this syndrome has severe forms of expression, including central nervous system abnormalities, cardiovascular disease, and gastrointestinal malformations, patients with this syndrome usually die within several weeks. To the best of our knowledge, no report yet has discussed the lifespan of AOS patients who do not evidence lethal anomalies.
CASE REPORT
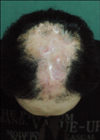
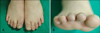
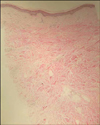
A 21-year-old man presented with a hairless atrophic patch (Fig. 1), CMTC on the dorsum of both hands, shortening of the fingers and toes, interdigital webs of the 2nd and 3rd toes, and total absence of the right 3rd toenail (Fig. 2). He was born at term and presented with small hairless patches over his scalp and total absence of the right 3rd toenail. The patient reported no history of maternal drug intake, infection, or radiation exposure during the pregnancy and no familial history of AOS, mental retardation, or CNS abnormalities. The scalp had a well-demarcated 10×12 cm hairless atrophic patch at its vertex. Histopathological examination of the scalp lesion revealed rete ridge flattening, collagen bundle thickening in the dermis, and the loss of appendages (Fig. 3). The skull x-ray showed a thinning of the bone underlying the hairless atrophic patch. No abnormalities were noted on brain computed tomography (CT) or on chest x-ray. The combination of aplasia cutis congenita and skeletal defects of the extremities resulted in a diagnosis of AOS. Gastrointestinal and urologic system examinations were recommended to the patient, but he refused to undergo them because he had already survived for more than 20 years with AOS.
DISCUSSION
AOS is a congenital disease characterized by aplasia cutis congenita, transverse limb defects, and CMTC1,2. It was initially described in 1945 by Adams and Oliver1. The disease largely has an autosomal dominant mode of inheritance, although autosomal recessive and sporadic modes often occur as well3,4. Our patient had no family history of congenital scalp or limb defects. Therefore, this case was likely a sporadic one.
The limb defects, the most common feature of this disease, are usually asymmetric. The lower limbs are more susceptible than the upper limbs. The range of involvement includes brachydactyly, syndactyly, agenesis of fingernails or toenails, loss of the terminal phalanges, or even more severe defects such as the complete absence of a finger, toe, hand, foot, or limb5,6. Our patient had interdigital webs of the 2nd and 3rd toes and absence of the 3rd toenail on the right. Scalp defects are the second most frequent finding in AOS4. The scalp lesions occasionally extend through the skull deformities. Our patient exhibited only a scalp defect and skull thinning. The lesion was confirmed as aplasia cutis congenita upon histopathological examination. Other associated defects include CMTC, central nervous system and cardiovascular malformations, accessory nipples, microphthalmia, and cleft lip7,8. Among these defects, our patient exhibited only CMTC.
Although the exact pathogenesis of AOS remains unknown, vascular impairment during embryogenesis has been proposed as a possible mechanism. In order to identify the genetic cause of AOS, Verdyck et al.9 evaluated a family with 10 affected individuals over four generations. However, they failed to find any disease-causing mutations.
Various expressions of AOS have been reported. This report shows that AOS without major organ abnormalities does not necessarily alter the normal lifespan. Additionally, if a newborn presents with aplasia cutis congenita and limb defects, dermatologists should consider AOS by conducting an evaluation of the central nervous system and looking for cardiovascular, gastrointestinal, and genitourinary malformations.
 XML Download
XML Download