

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
It is known that about one-third of deaths worldwide (i.e., ~17.3 million per year) are caused by cardiovascular dysfunctions [1]. Among patients with cardiovascular disease, 30.7% of patients were found to have both hypertension and dyslipidemia, and 66.3% of patients with diabetes had concomitant hypertension and dyslipidemia, in a 3-year retrospective study [2]. Thus, multiple drug therapy has been widely practiced for the treatment of problems including hypertension and dyslipidemia. Rosuvastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor, is commonly used in combination with telmisartan, an angiotensin II type-I receptor antagonist (ARB). Recently, Son et al. [3] reported increased pharmacokinetic (PK) exposure of rosuvastatin when coadministered with telmisartan. The absorption of rosuvastatin was accelerated (Cmax was doubled, with Tmax change from 5 h to 0.75 h.), whereas its AUC increased by only 1.18-fold when coadministered with telmisartan, although the cause of this phenomenon was not identified by the authors. In addition to the rosuvastatin–telmisartan interaction, it is well-known that cyclosporine also increases rosuvastatin AUC and Cmax by 1.5- and 3.0-fold and more severe interaction was reported in transplant recipients (>6 months, stable heart transplant recipients): the AUC and Cmax ratios of rosuvastatin (with/without cyclosporine) were of 7.1- and 10.6-fold, respectively [45]. Cyclosporine is an inhibitor of many transporters including OATP1B1/1B3, NTCP, and BCRP transporters and the rosuvastatin–cyclosporine interaction is known to be mediated by transporters.
Telmisartan prevents angiotensin II from exerting its vasoconstrictive effects on blood vessels, and thus has been widely used for the treatment of hypertension [67]. It has been reported that telmisartan is rapidly absorbed in the gastrointestinal tract (Tmax 0.75 h) [3] and is predominantly transported into the liver by organic anion transporting polypeptide (OATP) 1B3 [7], to be metabolized to an acylglucuronide conjugate by hepatic UDPglucuronosyltransferase (UGT) 1A3 [8], such that no unchanged parent molecule is excreted [9]. Recently, Hirano et al. [10] reported that telmisartan potently inhibited OATP1B1-mediated pravastatin uptake (inhibitory constant (Ki), 0.436 µM) and OATP1B3-mediated dioscin uptake (Ki, 1.03 µM) in vitro [11]. In addition to uptake transporter inhibition, telmisartan is also known to be a non-specific inhibitor of ATP-binding cassette (ABC) transporters, such as P-glycoprotein (P-gp), breast cancer resistance protein (BCRP) [12], and multidrug resistance-associated protein 2 (MPR2) [13].
Rosuvastatin, a highly selective and competitive HMG-CoA reductase inhibitor, reduces LDL and increases HDL cholesterol levels in blood [14]. It is mainly excreted unchanged into bile [1516], and less than 10% is metabolized to N-desmethylrosuvastatin by CYP2C9 [17]. Rosuvastatin is extensively distributed into the liver, presumably due to active uptake by OATP1B1 and OATP1B3, as well as by the sodium-dependent taurocholated co-transporting polypeptide (NTCP) transporters [181920], despite its low passive diffusion into hepatocytes [41521]. It is also a substrate of both liver canalicular and intestinal BCRP efflux transporters [212223].
Nowadays, it has been reported that the increased rosuvastatin exposure by concomitant medication is mediated by the BCRP transporter, rather than by OATP1B1/1B3 transporters [2425]. The aim of this study is to explore the mechanism of the transport-mediated interaction between rosuvastatin and two perpetrators, telmisartan and cyclosporine. Although many researchers have reported that PK changes of rosuvastatin are mainly due to OATP1B1-mediated interactions, we demonstrate here that alterations in the PK of rosuvastatin upon coadministration with telmisartan are mainly mediated by their sharing BCRP, not OATP1B1/1B3, transporters, using a physiologically based PK (PBPK) model. We also describe rosuvastatin and cyclosporine DDI with PBPK model focusing on transporter-mediated interactions. Moreover, because Asians show higher systemic exposure of rosuvastatin than Caucasians do [2627], the PBPK model reflecting exposures in Koreans was applied to the rosuvastatin model. PBPK modeling provides a bottom-up approach to PK predictions with integration of diverse information on human physiology, physiochemical properties of drugs, and drug metabolizing enzymes and transporters. Simcyp (version 15 release 1, Certara, Sheffied, UK) was used in this report to explore the contributions of BCRP and OATP1B1/1B3 transporters to drug interactions and to predict the PK of rosuvastatin.
A part of this paper was presented at the 11th international ISSX meeting (2016). A part of this study was first published as an original article in Biopharm Drug Dispos 2017; doi: 10.1002/bdd.2060, but was retracted by the authors because trivial miscalculations incurred by one misused input parameter were found throughout the simulated values in the article. For further details, please read doi: 10.1002/bdd.2082.
METHODS
Development of the telmisartan model
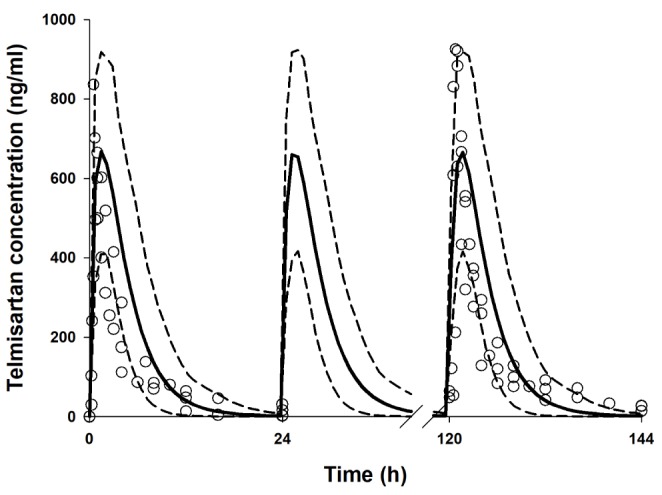
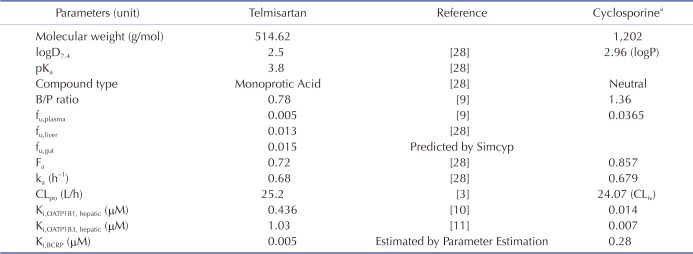
Simcyp (version 15 release 1, Certara, Sheffield, UK) was used to simulate and predict the time course of rosuvastatin in plasma with or without telmisartan. The telmisartan model was developed based on the report by Li et al. [28], where many of the physiochemical properties and in vitro PK parameters of telmisartan used herein are listed in Table 1. The simulation of telmisartan plasma concentration–time profiles was performed using a full-PBPK model of first-order absorption. The ka and Fa values are from Li et al. [28], and the nominal flow in gut model (Qgut), which reflects both permeability through the enterocyte membrane and villous blood flow [29], was estimated by Simcyp to be 15.564 L/h based on telmisartan physiochemical properties. The human PET data-derived kp values [28] for adipose tissue, bone, brain, gut, heart, kidney, lung, muscle, skin, and spleen used in this study were 0.35, 0.47, 0.25, 1.67, 1.54, 4.4, 1.07, 0.40, 0.26, and 0.41, respectively. However, we used the value reported by Rodgers and Rowland (0.12) [2830] for the hepatic kp because PET cannot discern telmisartan in hepatocytes from its metabolite, telmisartan-glucuronide, whose concentration is much higher than that of the parent compound in the liver [28]. The steadystate volume of distribution (Vss) was then estimated as 0.41 L/kg based on the aforementioned kp values using mathematical model 2 implemented in Simcyp [28]. For the telmisartan CLpo, the mean value reported in healthy subjects (25.2 L/h) was used [3].
Before the evaluation of telmisartan–rosuvastatin pharmacokinetic interactions, a clinical trial simulation of telmisartan was performed with virtual healthy populations (aged 20–50, female ratio of 0.5) in 10 trials of 10 subjects each. Simulated PK parameters and concentration–time profiles of telmisartan after a single and multiple (80 mg) oral administration were compared with observed PK data for validation.
Modification of the rosuvastatin model
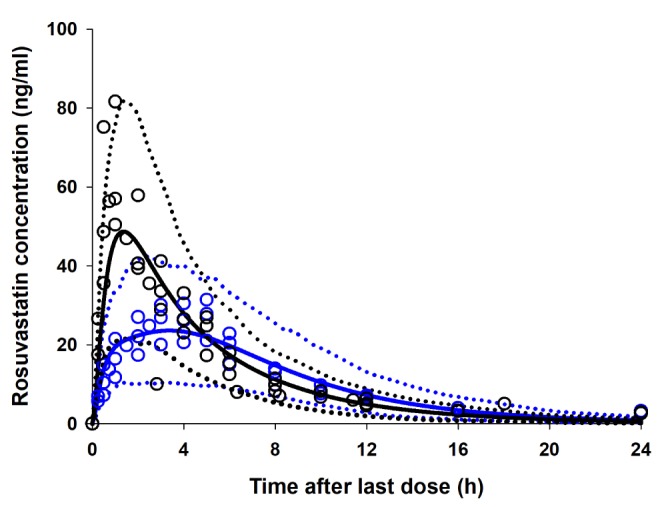
A PBPK model of rosuvastatin in a European Caucasian healthy volunteer population was previously constructed in Simcyp's drug library. Because Asians show higher systemic exposure of rosuvastatin than do Caucasians [26], the recommended starting dose for Asians (5 mg) is lower than that for Caucasians (10 mg) [27]. Although the cause of these racial differences is unclear, one possible explanation is that there may be differences in the activity or expression levels of uptake transporters of rosuvastatin, such as OATP1B1, OATP1B3, and NTCP, and/or an efflux transporter, such as BCRP. Because the PK data of a telmisartan–rosuvastatin interaction study conducted in Korean healthy volunteers were used as the reference [3], the rosuvastatin model in Simcyp's drug library was modified to reflect PK profiles in Koreans. Using Sensitivity Analysis (SA) and Parameter Estimation (PE) modules, the hepatic uptake clearance values of OATP1B1, OATP1B3, and NTCP for rosuvastatin were modified step-by-step to recover the published mean plasma concentrationtime profile of rosuvastatin in Korean populations [33132] from 5 different studies, whose data were obtained by digitization (Plot Digitizer, version 2.6.6, http://plotdigitizer.sourceforge.net). After these modifications, rosuvastatin concentrations in 10 virtual trials (10 healthy subjects per trial, aged 20–50, male/female=1:1) after a single (20 mg) oral dose were simulated in Simcyp.
Evaluation of the effects of telmisartan on the PK of rosuvastatin using PBPK modeling
Estimation of Ki,BCRP,intestine of telmisartan
Based on the above telmisartan and rosuvastatin models, telmisartan–rosuvastatin interaction was predicted. The in vitro inhibitory constants of telmisartan against hepatic OATP1B1 (Ki,OATP1B1,hepatic) and OATP1B3 (Ki,OATP1B3,hepatic) have been reported as 0.436 µM and 1.03 µM, respectively [1011]. Because there was no published Ki value of telmisartan against the intestinal BCRP transporter (Ki,BCRP,intestine), several methods were used to estimate the proper Ki,BCRP,intestine value of telmasartan; 1) its half-maximal inhibitory concentration (IC50,BCRP) [13] was used to calculate Ki,BCRP,intestine, by Eq. (1) described in [33]:
where S is substrate concentration and Km represents the Michaelis–Menten constant of the substrate. If S is near Km values, then Ki,BCRP,intestine would be IC50/2. 2) In addition to using the estimated Ki,BCRP,intestine value as IC50/2, a sensitivity analysis (SA) comparing simulated and observed Cmax and AUC ratios at varying Ki,BCRP,intestine estimates was conducted. 3) Based on the Ki,BCRP,intestine estimate obtained from SA, the parameter estimation (PE) module was used for the estimation of the Ki,BCRP,intestine value.
Simulation of clinical study
Clinical trials in 100 virtual healthy subjects (10 trials×10 subjects per trial, aged 20–50, male:female=1:1) were simulated in Simcyp with a dosing scheme identical to that reported by Son et al. [3]. After rosuvastatin (20 mg qd) was given alone or together with telmisartan (80 mg qd) for six days, plasma concentration profiles of rosuvastatin were simulated to compare with those reported elsewhere [332] (Table 2). The profiles of rosuvastatin in the gut and liver were also predicted to elucidate the mechanisms of interaction.
Evaluation of the effects of cyclosporine on the PK of rosuvastatin using PBPK modeling
The cyclosporine model reported by Jamei et al. [4] was used (Table 1). We validated the model using the data digitized from two different human studies [3435] (Plot Digitizer, version 2.6.6, http://plotdigitizer.sourceforge.net). Clinical trials in 100 virtual healthy subjects (10 trials×10 subjects per trial, aged 20–50, male:female=1:1) were simulated for the evaluation of the effects of cyclosporine on rosuvastatin PK. The profiles of rosuvastatin (20 mg) PK were simulated with or without cyclosporine (200 mg, BID).
RESULTS
Development of the telmisartan model
Telmisartan plasma concentration–time profiles after single oral administration (80 mg) were simulated using its physicochemical parameters and PK parameters (Table 1). The Cmax, AUC, and Tmax after administration of telmisartan 80 mg predicted by Simcyp (10 trials of 10 subjects each) were similar to the published data [63637] and the simulation results of multiple administrations of telmisartan 80 mg for 6 days also well explained the observed data (Fig. 1) [33839].
Modification of the rosuvastatin model
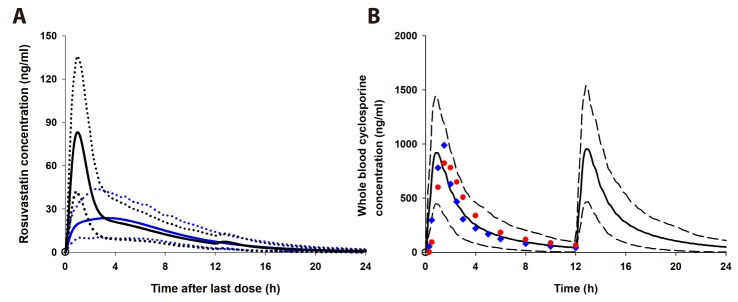
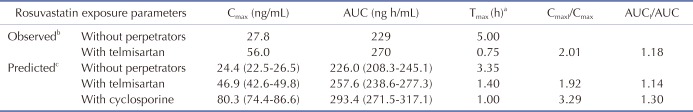
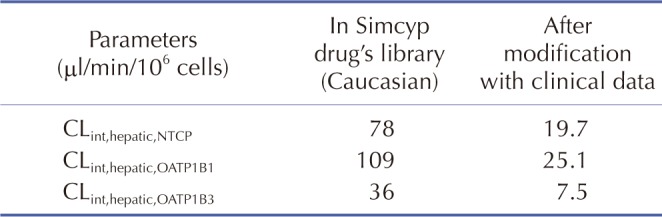
The rosuvastatin model in the Simcyp drug library was modified by the authors to reproduce the Cmax and AUC in Koreans, which are about twice as high as in Caucasians [1940]. The total CLint, uptake transporters value estimated using the SA method was adjusted for the ratio of CLint,hepatic,NTCP, CLint,hepatic,OATP1B1, and CLint,hepatic,OATP1B3 in Caucasians in Simcyp. The final CLint values were determined by the PE method using observed data from 5 different studies in Koreans. Upon comparing the PK data reported from Koreans [33132] with our Simcyp-simulated ones, the CLint values of the hepatic uptake transporters of rosuvastatin were adjusted (Table 3): they were about 1/5 of CLints of European Caucasians (Pre-existing CLint,hepatic,NTCP, CLint,hepatic,OATP1B1, and CLint,hepatic,OATP1B3 values of 78, 109, and 36 µl/min/106 cells in Simcyp were modified as 19.7, 25.1, and 7.5 µl/min/106 cells, respectively, to be used for our simulation.). The Cmax, AUC, and Tmax of rosuvastatin (20 mg single oral dosing) simulated using the modified CLints of uptake transporters were 25.7±10.1 ng/ml, 235±95.2 ng h/ml, and 3.30 (1.92–3.85) h, respectively (Fig. 2).
Evaluation of the effect of telmisartan on the PK of rosuvastatin using PBPK modeling
Prior to conducting the simulation of drug–drug interaction studies, the Ki,BCRP,intestine of telmisartan was estimated. We initially tried IC50,BCRP/2 (8.45 µM) using Eq. (1) as the Ki,BCRP,intestine of telmisartan. However, the AUCI/AUC and CmaxI/Cmax of rosuvastatin (with/without telmisartan) predicted thereby were both 1.00-fold which implied that the calculated Ki,BCRP,intestine (8.45 µM) as half the IC50,BCRP (16.9 µM) [13] under-predicted the magnitude of the rosuvastatin–telmisartan interaction. Thus, we carried out SA of Ki,BCRP,intestine. The Ki,BCRP,intestine chosen from SA (0.003 µM) was much lower than 8.45 µM, that obtained from IC50,BCRP and this was used as the as the initial value for the PE module. The final estimate of Ki,BCRP,intestine obtained from the PE module (0.005 µM) best explained the observed drug interactions. Thus, this Ki,BCRP,intestine value was chosen to be employed in the rosuvastatin–telmisartan interaction study. Assuming that the affinity of BCRP present in the liver and intestine are the same, we also implemented this value to account for the possible inhibition of BCRP transporter existing in hepatocytes.
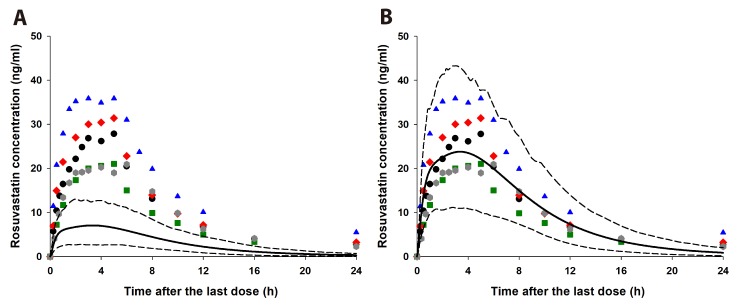
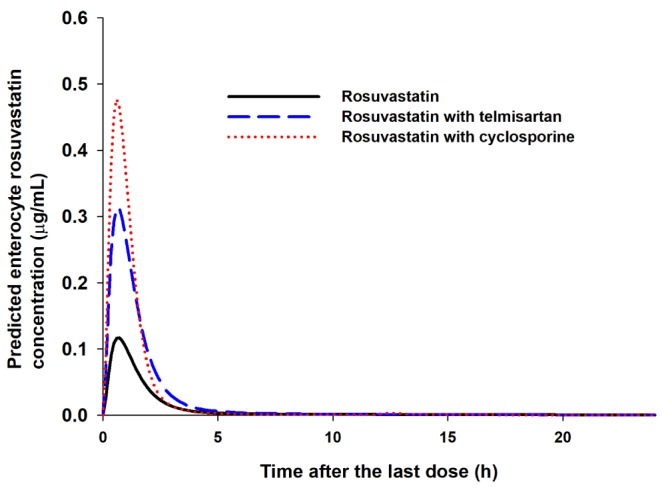
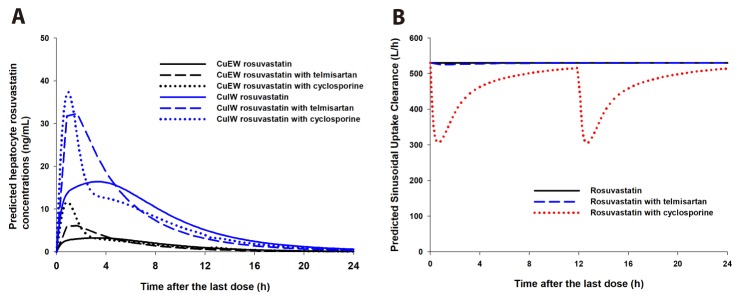
After the simulation of clinical studies, the predicted changes in plasma concentration–time profiles of rosuvastatin upon coadministration of telmisartan are overlaid with reported data in Fig. 3. The observed and predicted Cmax, Tmax, and AUC of 20 mg rosuvastatin with or without coadministration of 80 mg telmisartan are listed in Table 2. The predicted AUCI/AUC (1.14) and CmaxI/Cmax (1.92) of rosuvastatin were similar to those reported by Son et al. (AUCI/AUC 1.18 and CmaxI/Cmax 2.01) [3]. The changes in gut and liver rosuvastatin concentrations were predicted to elucidate the intestinal BCRP-mediated interactions. In the GI tract, telmisartan increased the enterocyte concentration of rosuvastatin by 2.20-fold (Fig. 4, the representative intestinal segment, jejunum), that indicated the inhibition of BCRP efflux transporter by telmisartan. Extracellular and intracellular concentrations of rosuvastatin in liver were also available in these models (Fig. 5). Changes in the extracellular and intracellular rosuvastatin concentration-time profiles of rosuvastatin by telmisartan were almost identical (accelerated Tmax and increased Cmax), thus the extracellular/ intracellular AUC ratio of rosuvastatin did not change at all (0.203 without telmisartan versus 0.201 with telmisartan). Moreover, the change in sinusoidal uptake clearance of rosuvastatin is insignificant. These results also imply that the hepatic transporter-mediated interaction by telmisartan is negligible.
Evaluation of the effect of cyclosporine on the PK of rosuvastatin using PBPK modeling
The simulated whole blood concentration–time profiles of cyclosporine after 200 mg BID oral administration well explained the observed data (Fig. 6B). The predicted changes of plasma exposures of rosuvastatin are shown in Fig. 6A and the PK parameters are listed in Table 2. The changes in gut and liver rosuvastatin concentrations were also estimated in this simulation. The predicted enterocyte concentrations of rosuvastatin in the presence of cyclosporine were 3.27-fold higher than those of rosuvastatin alone. In hepatocytes, the extracellular/intracellular AUC ratio of rosuvastatin was predicted to increase by 22.7% by cyclosporine (0.203 without cyclosporine versus 0.249 with cyclosporine) and this implied that both of BCRP and OATP1B1/3-mediated interactions by cyclosporine play an important role in rosuvastatin disposition. The change in sinusoidal uptake clearance of rosuvastatin by cyclosporine appeared significant compared to that by telmisartan (Fig. 5B).
DISCUSSION
This study primarily aimed to demonstrate that the BCRP-mediated drug interactions are critical in rosuvastatin by presenting two example cases. The first case is the rosuvastatin–telmisartan study. The primacy of the contribution of the intestinal BCRP transporter seems to be in accord with the PK characteristics of telmisartan and rosuvastatin. Telmisartan (Tmax 0.75 h) is absorbed faster than rosuvastatin (Tmax 5 h), presumably due to its highly permeable absorption in the GI tract. The Simcyp-predicted intestinal tissue concentration of telmisartan multiplied by fu,gut (0.015) increased above its Ki,BCRP,intestine (0.005 µM) immediately after administration, and was maintained for about 9 h (data not shown). In other words, Simcyp predicted that telmisartan pre-occupied the binding sites of intestinal BCRP owing to its faster absorption characteristic, before rosuvastatin molecules could engage with the BCRP transporters, resulting in its accelerated absorption (Cmax↑, Tmax↓) without evident decrease in systemic clearance. Mixed-effects modeling results also supported that the rosuvastatin population PK parameter changes by telmisartan implied a modest increase (1.3-fold) in relative bioavailability despite the drastic acceleration of the absorption rate [32].
The second case is the rosuvastatin–cyclosporine study which supports the view that both of the BCRP and OATP1B1/3 transporters contribute to the increased exposure of rosuvastatin. Unlike the rosuvastatin–telmisartan case, hepatic OATP1B1/3-mediated interactions were crucial because cyclosporine exhibited more potent inhibitory effects (Ki, OATP1B1=0.014 µM, Ki,OATP1B3=0.007 µM) against OATP1B1/3 than telmisartan did (Ki,OATP1B1=0.436 µM, Ki, OATP1B3=1.03 µM). This enabled cyclosporine to reach concentrations sufficient to inhibit OATP1B1/3 at the target site. Reproduced cyclosporine PBPK model well explained the observed data and the simulation results were also accordant with those in Jamei's report [4].
The changes in gut and liver rosuvastatin concentration profiles were also predicted in our study. Using the Advanced Dissolution, Absorption and Metabolism (ADAM) model implemented in Simcyp, changes in rosuvastatin concentration in each segment were predicted. In the representative intestinal segment (jejunum), one of the segments where the BCRP transporters are abundantly expressed [41], the rosuvastatin exposure in enterocytes was 2.20-fold and 3.27-fold higher in the presence of telmisartan and cyclosporine, respectively (Fig. 4, patterns in the other segments were similar: data not shown), supporting the hypothesis that the BCRP transporter is a major cause of drug interactions of rosuvastatin in both cases.
Increased absorption of rosuvastatin by telmisartan and cyclosporine has driven a greater fraction of rosuvastatin to reach the liver and caused accelerated Tmax and increased Cmax in both extracellular water and intracellular water of hepatocytes. However, there are critical differences in two cases. In the case of telmisartan interactions, it is of note is that the hepatic uptake of rosuvastatin was increased by telmisartan without a change in extracellular water/intracellular water rosuvastatin AUCs, despite its known inhibition of OATP1B1/1B3 transporters (Ki,OATP1B1/OATP1B3,hepatic values of 0.436 and 1.03 µM, respectively) [1011]. On the contrary, in the case of cyclosporine interactions, extracellular/ intracellular rosuvastatin AUC ratios in hepatocytes increased by 22.7% in the presence of cyclosporine, which indicated that rosuvastatin uptake into the hepatocyte was reduced. Similarly, predicted sinusoidal uptake clearance of rosuvastatin did not change in the presence of telmisartan whereas that of rosuvastatin was reduced in the presence of cyclosporine (Fig. 5B).
There is another report supporting this inference. Gemfibrozil inhibits OATP1B1-, OATP2B1-, and NTCP-mediated rosuvastatin uptake [20]. In a gemfibrozil–rosuvastatin interaction study in humans, the AUC and Cmax of rosuvastatin were doubled, whereas those of the metabolite, N-desmethyl rosuvastatin, were halved [42]. This implies that gemfibrozil inhibited rosuvastatin uptake into hepatocytes, resulting in the decreased N-desmethylation of rosuvastatin by CYP2C9 in hepatocytes. On the other hand, the AUC and Cmax of N-desmethyl rosuvastatin increased by 1.07- and 1.53- fold, respectively, with coadministration of telmisartan in Son's report [3], which was the opposite of the results of the gemfibrozil-rosuvastatin study. Thus, the mechanistic explanation of the interaction between rosuvastatin and telmisartan proposed in this report is: telmisartan inhibits rosuvastatin efflux in the GI tract → the amount of rosuvastatin absorbed into the portal vein is increased → the hepatic uptake of rosuvastatin is increased.
To determine the Ki,BCRP,intestine value of telmisartan, we employed a top-down approach using data from five different clinical DDI studies. It is known that some discrepancy exists between in vitro and in vivo Ki values as exemplified by Kato et al. [43]. Because the in vitro Ki,BCRP,intestine value under-predicted the effect of telmisartan on the rosuvastatin exposures observed in clinical trials, we decided to use a top-down approach. Hu and his colleagues performed an in vitro transporter interaction study with telmisartan and rosuvastatin [25]. Their result was that the efflux ratio of rosuvastatin significantly decreased by 42% by 1 µM of telmisartan and this was consistent with our results.
We used in vivo CLpo rather than metabolic clearance in hepatocytes (CLint,met) for the telmisartan CL in the simulation step for the following reasons: 1) an in vivo CLpo value (25.2 L/h) was suitable to recover the observed telmisartan PK; 2) the metabolic pathway of telmisartan in hepatocytes did not overlap that of rosuvastatin; and 3) the half-life of telmisartan is over 20 h [9] despite the CLint,met of telmisartan being reported as 395 µl/min/ mg microsomal protein [44] or 1,210 µl/min/mg microsomal protein [28] belonging to high extraction ratio drugs. The discrepancy between CLint,met and in vivo CLpo can be explained by the enterohepatic circulation of telmisartan-glucuronide after biliary excretion and deconjugation in the intestine [828]. Failure to reflect the reabsorbed portion of telmisartan can result in overestimation of CLint,all, leading to overestimation of CLH, and ultimately, of CLin vivo. Because there is no quantitative information on biliary-excreted and recirculated telmisartan and its glucuronide, our model did not address the enterohepatic recirculation of telmisartan.
To reproduce rosuvastatin exposure in healthy Korean subjects, the CLint values of uptake transporters were modified. In Asian subjects, the exposure of rosuvastatin is known to be almost twice that in Caucasians [27]. This has been suggested to be due to a race-related difference in CLH, but not in CLR or FaFg [45]. OATP1B1 (SLCO1B1) has single-nucleotide polymorphisms (SNPs) of T521>C (Val174Ala) and A388>G (Asn130Asp) associated with the OATP1B1 *1a (174Val and 130Asn), *1b (174Val and 130Asp), *5 (174Ala and 130Asn), and *15 (174Ala and 130Asp) alleles [46], which cause genetic variability in rosuvastatin PK. However, it does not explain the race-related variability of rosuvastatin PK. The allelic frequency of 521T<C, which is closely correlated with rosuvastatin PK, is similar between Asians and Caucasians [2147]. Despite the SLCO1B1 genotype being identical between Caucasians and Chinese, the Cmax and AUC of Chinese were approximately twice those of Caucasians [2140]. Based on these reports, Tomita et al. [45] suggested that there may be a difference in protein expression levels of hepatic OATP1B1 between Asians and Caucasians. The effect of the genetic polymorphism of OATP1B3 and NTCP on the racial differences in rosuvastatin PK is unclear. Similar to the case of rosuvastatin, pravastatin, another substrate of OATP1B1, showed higher exposure in Asians as well. Li et al. tried to explain this with the OATP1B1 genetic variant, however, more supporting evidences are needed to apply their assertion for PBPK models [48].
A few reports [404950] suggested that the genetic polymorphism of BCRP may have caused inter-racial difference in rosuvastatin exposure, but more studies are needed to conclude this. Considering the degree of uncertainty with respect to uptake transporter-mediated racial differences, CLint values of hepatic uptake transporters had to be adjusted based on the observed rosuvastatin systemic exposures, which is a limitation of our model. Therefore, quantitative prediction of drug interaction potential for telmisartan as an intestinal BCRP inhibitor in this report may be reproduced in other races if appropriate clinical and/or in vitro data are available.
In conclusion, our PBPK modeling to evaluate rosuvastatin–telmisartan/cyclosporine interaction revealed that the intestinal BCRP transporter is a major contributor to the interactions.
 XML Download
XML Download