

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acetaminophen (APAP), also known as paracetamol and N-acetylp- aminophenolis, is the most widely used analgesic and antipyretic agent in the world with an excellent safety profile when administered at therapeutic doses [1]. However, APAP in high doses can cause hepatotoxicity and is recognized as a major cause of acute liver failure (ALF). In the USA and Europe, acetaminophen overdose cause hepatotoxicity with over 300,000 hospitalizations annually and up to 42% of all cases of ALF attributable to acetaminophen overdose [234]. Mechanistically, APAP is metabolized by the cytochrome P450 system into an electrophilic metabolite, N-acetyl-p-amino-benzoquinone imine (NAPQI) which reacts with glutathione (GSH) and leads to a profound depletion of hepatocellular GSH. Subsequently, NAPQI binds to mitochondrial proteins, which in turn causes oxidative stress, increased phosphorylation and adenosine triphosphate (ATP) depletion that may trigger signaling pathways through mitochondrial toxicity, leading to DNA damage and liver injury [56].
Although the precise mechanism by which APAP or its metabolites cause liver injury is still unknown, cell death and organ failure most likely result from the cumulative and additive effects from oxidative damage and redox balances. The nuclear factor-like 2 (Nrf2), and its repressor kelch-like ECH-associated protein 1 (Keap1), have been characterized as an important endogenous cellular mechanism for coping with oxidative stress [7]. Nrf2 belongs to the basic leucine zipper (bZip) transcription factor family and has been implicated as a key molecule involved in antioxidant-responsive element (ARE)-mediated gene expression [8]. Under basal conditions, Nrf2-mediated transcription is turned off because of the inhibitory effect of Keap1. Keap1 binds to Nrf2 and sequesters the molecule from nuclei, preventing Nrf2 from activating target genes. Cullin 3 (Cul3), one of the components of ubiquitin ligase, interacts with Keap1 and mediates the degradation of Nrf2 through ubiquitin-proteasome proteolytic pathway [9]. It has been reported that Nrf2 plays a protective role against APAP hepatotoxicity by regulating both drug metabolizing enzymes and antioxidant genes through the ARE [10]. In addition, mice lacking the Nrf2 transcription factor had difficulty in detoxifying APAP and its metabolites because of the lower level of hepatic GSH [11].
Carnosic acid (CA), a naturally occurring catechol type polyphenolic diterpene present in rosemary (Rosmarinus officinalis; Lamiaceae), has a wide array of pharmacological properties such as anti-oxidative, neuroprotective, anti-nephrotoxicity, anti-inf lammatory and anticarcinogenic activities [12131415]. Recent study has shown that CA prevents lipid accumulation in hepatocytes through the EGFR/MAPK pathway [16]. It was also reported that CA attenuated lipopolysaccharide-induced liver injury in rats via fortifying cellular antioxidant defense system [17]. Importantly, CA was found to stimulate Keap1/Nrf2 signaling, thus resulting in the production of antioxidants such as GSH, thereby reducing oxidative stress [18].
In this study, we investigated the protective effect of CA on APAP-induced hepatotoxicity and its underlying mechanism in mice. The results indicated that CA ameliorated APAP-induced acute liver injury through regulation of anti-oxidant system. Further examination of the signaling pathways indicated that CA facilitated Nrf2 translocation into nuclear and upregulated antioxidant genes expression. Our findings suggest that CA has protective effect on APAP-induced hepatotoxicity through regulation of antioxidant system.
METHODS
Material
Acetaminophen, Carnosic acid, Sodium carboxymethyl cellulose, Hematoxylin and Eosin were purchased from Sigma Aldrich (St. Louis, MO, USA). Anti-Cleaved caspase 3, anti-Bax, anti-Bcl-2, anti-phospho-JNK, anti-JNK, anti-Nrf2, anti-Lamin B1, anti-IκBα, anti-p65, anti-phospho-IκBα, anti-phospho-p65, anti-Keap1, anti-Cul3 and anti-β-actin were purchased from Cell Signaling Technology (Beverly, MA, USA). Trizol reagent and SYBR® Green PCR Master Mix were purchased from Invitrogen (Carlsbad, CA, USA).
Animals and treatment
Male C57BL/6J mice (8 weeks old) were purchased from Experimental Animal Center of Henan Province (Henan, China) and maintained with free access to pellet food and water in specific-pathogen-free (SPF) facility with a 12 h light/dark cycle. Animal welfare and experimental procedures were followed in accordance with the Guide for Care and Use of Laboratory Animals and the related ethical regulations of Zhengzhou University Animal Care and Use Committee. CA was suspended in 0.5% carboxymethylcellulose sodium (CMC-Na) solution and administered (100 mg/kg per day) by gavage once daily for three days according to the previous reports [1719]. The vehicle group and APAP group were administered by 0.5% CMC-Na solution. To induce APAP hepatotoxicity, APAP was dissolved in a saline solution and administered by intraperitoneal (i.p.) injection at a single dose of 400 mg/kg. The vehicle group and CA alone group were injected i.p. with saline solution. All mice were sacrificed after APAP treatment for 6 h. A portion of the liver was fixed in 10% buffered formalin and the remaining liver tissues were flash frozen in liquid nitrogen and stored at -80℃ for further analysis.
Histological analysis
Liver samples were formalin-fixed, paraffin embedded, cut into sections (thickness: 5 µM) and stained with hematoxylin and eosin (H&E) following a standard protocol. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining in liver sections was performed with an In-Situ Cell Death Detection Kit according to the manufacturer's instructions (Roche Diagnostics, Indianapolis, IN). The images were visualized by microscopy (IX73, Olympus, Japan).
Determination of plasma ALT, AST and LDH concentrations
Aspartate aminotransferase (AST), alanine aminotransferase (ALT) and lactate dehydrogenase (LDH) were quantified in serum using a commercial kit. (Nanjing Jiancheng bioengineering institute, Nanjing, China).
Measurement of ROS, MDA and GSH in liver tissues
The levels of reactive oxygen species (ROS), malondialdehyde (MDA) and glutathione (GSH) were measured using a commercially available kit (Nanjing Jiancheng bioengineering institute, Nanjing, China). Briefly, liver weights were measured and homogenized in 2 ml ice-PBS. The samples were centrifuged at 2000 g for 10 minutes at 4℃. The MDA levels in the supernatant was determined by thiobarbituric acid (TBA) reaction and the absorbance was measured at 532 nm. The GSH levels were measured by the enzymatic recycling method using glutathione reductase and 5', 5'-dithio-bis (2-nitrobenzoic acid) (DTNB). 2-nitro-5-thiobenzoic acid (TNB) formation is monitored at 412 nm. ROS levels in liver tissues were determined using 2, 7-dichlorofluorescein diacetate (DCFH-DA) as a fluorescent probe. The supernatant (190 µl) was incubated with 1 mM DCFH-DA (10 µl) at 37℃ for 30 min. The fluorescence intensity was measured at an excitation and emission wavelength of 485 nm and 525 nm, respectively, using a spectrofluorometer (Hitachi, Tokyo, Japan).
Quantitative real-time polymerase chain reaction (PCR)
Isolation of total RNA from liver samples was performed using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. First-strand cDNA was synthesized from RNA using M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA). Real-time PCR was performed with the ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA) using SYBR® Green PCR Master Mix (Invitrogen, Carlsbad, CA). The primers were listed in Table 1. Relative gene expression levels were analyzed using the 2(-ΔΔCt) method and normalized against the expression of β-actin.
Western blot analysis
In Brief, liver samples were homogenized in RIPA buffer containing protease/phosphatase inhibitor cocktail (Cell Signaling Technology, Danvers, MA). Nuclei were isolated from mouse livers using NE-PER™ nuclear and cytoplasmic extraction reagents (Pierce, Rochford, IL) according to the manufacturer's instructions. The protein content was determined by a BCATM protein assay Kit (Pierce, Rochford, IL). Total cellular protein or nuclear protein were resolved by 10% SDS-PAGE and transferred onto a polyvinylidene diuoride membrane (Millipore, Bedford, MA). The membrane was blocked with 5% nonfat milk and incubated with primary antibody overnight at 4℃, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody (1:1000; Santa Cruz biotechnology, Santa Cruz, CA) for 1 h. The protein bands were visualized with enhanced chemiluminescence reagents (Pierce, Rochford, IL) using western blotting detection system according to the manufacturer's instructions. The densities of protein bands were determined by ImageJ software.
Co-immunoprecipitation (Co-IP) assay
Briefly, liver samples were homogenized in 1% Triton X-100 lysis buffer containing protease/phosphatase inhibitor cocktail (Cell Signaling Technology, Danvers, MA). After centrifugation at 12,000 g for 10 min, cell lysates were immunoprecipitated with Keap1 antibody with protein A/G-Sepharose. After incubation at 4℃ overnight, the beads were washed, separated by SDS-PAGE and analyzed by immunoblot with antibodies. The protein bands were visualized with enhanced chemiluminescence reagents (Pierce, Rochford, IL) using western blotting detection system according to the manufacturer's instructions. The densities of protein bands were determined by ImageJ software.
RESULTS
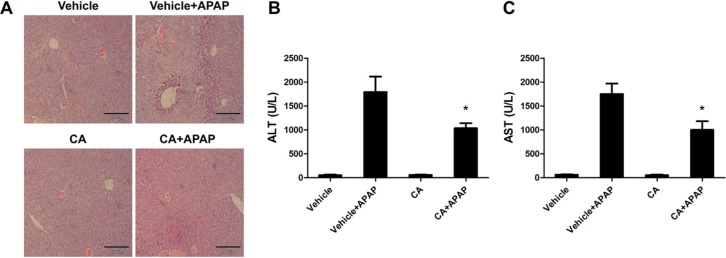
CA protected against APAP-induced liver injury
To investigate the effects of CA on the development of APAP-induced hepatotoxicity, mice were co-administrated with CA and APAP. Histological analysis of liver sections revealed that massive necroses and inflammation were present in APAP-treated mice. CA treatment significantly ameliorated APAP-induced liver injury (Fig. 1A). The serum levels of both AST and ALT, the markers of liver damage, were also markedly elevated by treatment with APAP. CA treatment significantly inhibited APAP-induced AST and ALT levels in the serum (Fig. 1B, C). These data indicated that CA protected against APAP-induced liver injury.
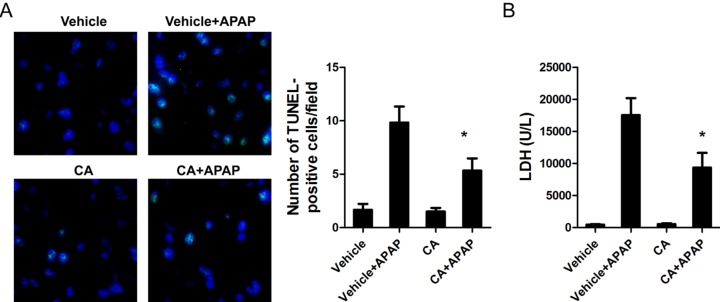
CA ameliorated APAP-induced cell death in the liver
DNA fragmentation, a characteristic feature of APAP-induced hepatocyte death, was determined by TUNEL assay in APAP-treated mice. As shown in Fig. 2A, APAP treatment caused DNA fragmentation in the liver and CA had strong protective effect on APAP-induced hepatocytes death. LDH releasing, as a marker for cell necrosis, is significantly reduced by CA treatment (Fig. 2B).
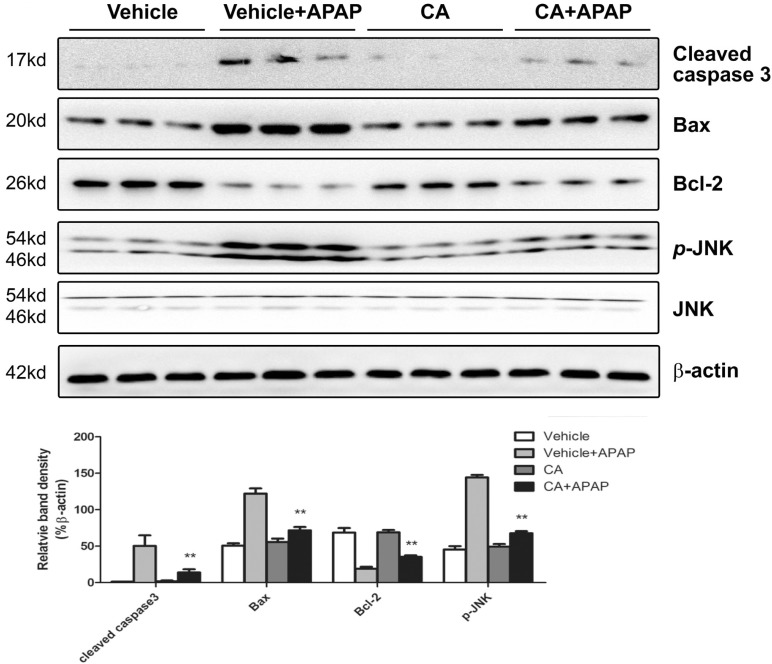
To confirm the effect of CA on APAP-induced cell death in the liver, we further determined the protein expression related to cell death. As shown in Fig. 3, APAP treatment increased Bax and cleaved caspase 3 expression and decreased Bcl-2 protein expression. However, the effects were reversed by CA treatment. The level of phosphorylated c-Jun N-terminal kinase (JNK), which has been documented to amplify APAP-induced mitochondrial oxidative stress, was significantly increasing in APAP-treated mice. CA treatment markedly reduced APAP-induced JNK phosphorylation. These data indicated that CA ameliorated APAP-induced cell death in the liver.
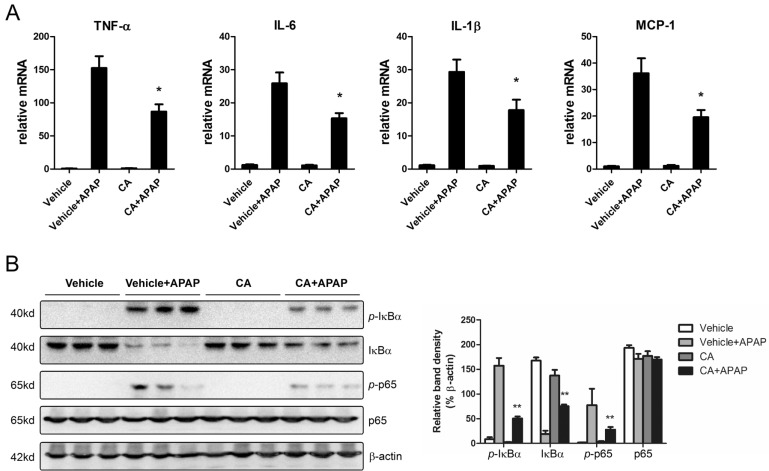
CA inhibited APAP-induced hepatic inflammation
APAP overdose-induced hepatocytes necrosis cause kuppfer cells activation in the liver. To investigate the effects of CA on hepatic inflammation, the hepatic mRNA expression levels of inflammatory cytokines were determined by real-time PCR. As shown in Fig. 4A, CA treatment significantly inhibited APAP-induced TNF-α, IL-1β, IL-6 and MCP-1 mRNA expression. NF-κB plays essential roles in transcriptional induction of those genes involved in inflammation [20]. Here, we assessed the effects of CA on NF-κB signaling pathway. Western blot results showed that CA significantly inhibited IκBα and p65 phosphorylation (Fig. 4B). The data indicated that CA might protect against APAP-induced hepatic inflammation through NF-κB signaling pathway.
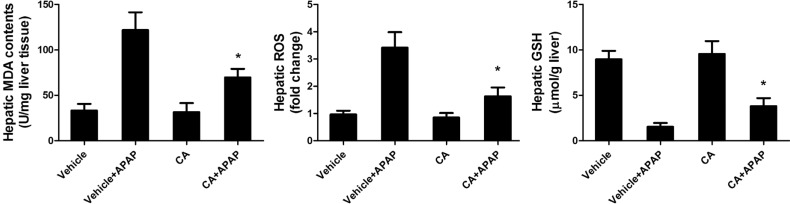
CA treatment reduced APAP-induced hepatic oxidative stress
In order to investigate the role of CA in APAP-induced oxidative injury, we investigated the antioxidant capacity of CA in liver tissue. As shown in Fig. 5, hepatic MDA contents were increasing after APAP treatment. CA inhibited APAP-induced hepatic MDA contents increasing. Moreover, CA decreased ROS accumulation in the liver compared with APAP alone group. In addition, we examined the total GSH to assess the detoxification status of the cells. Nevertheless, total GSH levels in APAP-injured liver increased by CA treatment. The result suggested that CA had a strong antioxidant capacity against APAP-induced liver injury.
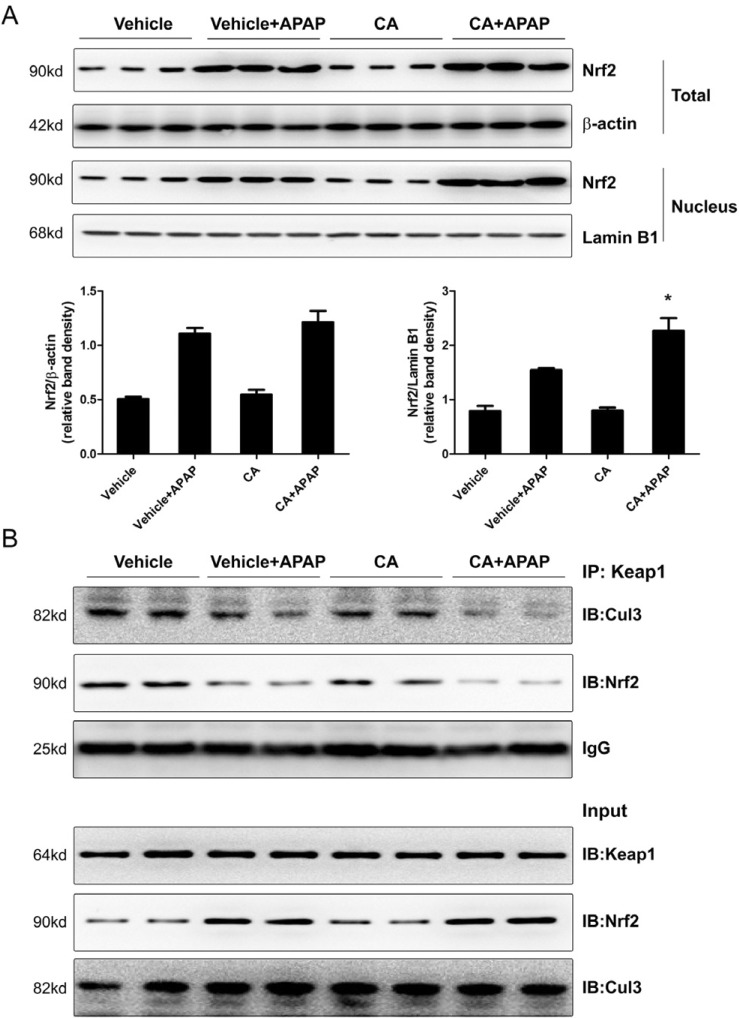
CA facilitated APAP-induced Nrf2 translocation into nuclear
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a master regulator for transcriptional activation of a wide spectrum of genes related to the antioxidant defense system. As shown in Fig. 6A, APAP alone treatment induced Nrf2 protein expression but not CA alone treatment. Interestingly, the level of Nrf2 protein in nuclear was higher in CA+APAP treatment group compared to APAP alone group. However, CA alone treatment did not induce Nrf2 translocation. It is known that Keap1 tethers Nrf2 in the cytoplasm and represses the transactivation activity of Nrf2 [9]. We further examined the effect of CA on interaction between Nrf2 and Keap1. As show in Fig. 6B, CO-IP assay showed that CA treatment facilitated dissociation of Nrf2 from Keap1. Moreover, CA treatment enhanced dissociation of Cul3 from Keap1. These data indicated that CA induced Nrf2 translocation into nuclear through facilitating dissociation of Nrf2 from Keap1.
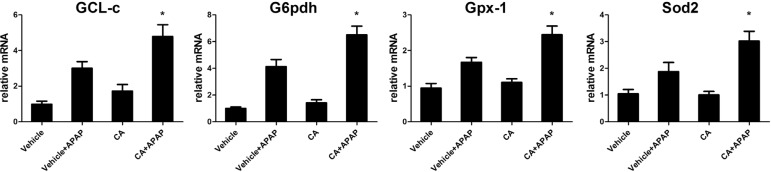
CA increased anti-oxidant genes expressions in APAPtreated mice
As excessive ROS accumulation occurs as a result of the imbalance between ROS generating and scavenging systems. It is well known that Nrf2 regulated anti-oxidant genes expression such as GCL-c, G6pdh, Gpx-1 and Sod2. We further examined these genes expression in the liver. As shown in Fig. 7, after APAP treatment, these genes expression increasing to against APAP-induced ROS accumulation. However, CA significantly increasing these genes expression compared with APAP alone group. Our data indicated that CA treatment-facilitated APAP-induced Nrf2 translocation resulted in anti-oxidant genes expression increasing.
DISCUSSION
In this study, we investigated the effects of CA on APAP-induced hepatotoxictiy in mice. We found that CA treatment significantly ameliorated APAP-induced hepatic necrosis and inflammation. Importantly, CA facilitated Nrf2 nuclear translocation, causing induction of Nrf2-dependent genes, which contributes to reduction of oxidative stress and protection from acetaminophen hepatotoxicity.
APAP overdose can cause severe hepatotoxicity and even liver failure in experimental animals and humans. Despite substantial efforts over the last 30 years, the mechanism of APAP-induced hepatotoxicity is still not completely understood [21]. However, numerous studies have demonstrated that oxidative stress plays a key role in APAP-induced hepatotoxicity [212223]. CA is a polyphenolic diterpene antioxidant found in rosemary plant prevents lipid peroxidation and lipopolysaccharide-induced liver injury via regulation of cellular antioxidant defense system [1724]. Moreover, CA (100 mg/kg) prevents isoproterenol-induced myocardial oxidative stress and apoptosis in mice [19]. It also has been reported that CA, as a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1 [18]. In addition, activated glutathione metabolism participates in protective effects of carnosic acid against oxidative stress in neuronal HT22 cells [25]. Based on the mechanism underlying antioxidant properties of CA, the present study was undertaken to know the protective effect of CA against APAP-induced oxidative stress and hepatotoxicity in mice. Our data showed that CA treatment decreased hepatic MDA contents and ROS accumulation (Fig. 5). It is well known that N-acetylcysteine (NAC), as an anti-oxidant, can protect against APAP-induced liver injury. Administration of NAC results in rapid GSH formation, which enhances the capacity to scavenge NAPQI and thus prevents all upstream-initiating events, such as protein binding and mitochondrial ROS formation [26]. CA administration increased GSH level in APAP-treated mice, suggesting that CA may block the upstream-initiating events.
In response to oxidative stress, the antioxidant defense system is often activated as a compensatory response to protect cell damage by maintaining cellular redox homeostasis [27]. The transcription factor Nrf2 regulates the basal and inducible expression of numerous detoxifying and antioxidant genes. Nrf2 has been shown to be actively involved in scavenging of APAP overdose-induced ROS in mice [28]. Our study showed that CA treatment facilitated Nrf2 translocation into nuclear and upregulated anti-oxidant genes mRNA expression (Fig. 6, 7). Moreover, we found that CA enhanced dissociation of Nrf2 from Keap1. CA treatment also facilitated dissociation of Cul3 from Keap1. It has been reported that CA binds to the BTB (Broad-Complex, Tramtrack and Bric-a-brac) and IVR (the intervening region) domains of Keap1 [18]. These two domains of Keap1 are required for Cul3 and Nrf2 bindings [29]. However, our data showed that CA alone treatment was not sufficient to block the interaction between Nrf2 and Keap1, indicating that CA could not bind to Keap1 under normal condition. APAP treatment induced exposure of the BTB and IVR domains of Keap1, which, in turn, might facilitate the binding of CA to Keap1. In addition, the binding of the APAP metabolite NAPQI to mitochondrial proteins trigger excessive accumulation of both ROS and peroxynitrite, which in turn cause ATP depletion and necrosis [30]. Furthermore, ROS can induce activation and phosphorylation of JNK, which exacerbates oxidative stress [31]. CA treatment significantly protected APAPinduced cell necrosis and JNK phosphorylation (Fig. 2, 3), suggesting that CA might inhibited APAP-induced cell necrosis and JNK activation through ameliorating oxidative stress.
Oxidative stress plays an important role in APAP-induced hepatotoxicity. In addition to inducing direct cellular damage, oxidants can activate transcription factors including NF-κB, which regulate the production of inflammatory mediators implicated in hepatotoxicity [32]. The necrotic hepatocytes induced by APAP release danger-associated- molecular patterns (DAMPs), which are recognized by resident hepatic macrophages and neutrophils, leading to the activation of these cells. Activated hepatic macrophages release various pro-inf lammatory cytokines, such as TNF-α or IL-1β, which enhance APAPinduced liver injury. Moreover, activated hepatic macrophages secret chemokines such as MCP-1 thereby further enhancing inflammation and increasing the influx of immune cells [33]. As an anti-oxidant compound, CA might protect against APAPinduced hepatocytes death through regulation of cellular antioxidant defense system. Our data showed that CA treatment significantly inhibited APAP-induced pro-inflammatory cytokines mRNA expression and NF-κB actvation in the liver (Fig. 4), suggesting that CA might suppress hepatic inflammation through protecting against APAP-induced hepatocyte death.
In summary, this study clearly demonstrated that CA protected against APAP-induced liver injury and enhanced antioxidant defense system through facilitating Nrf2 translocation into nuclear. Our study suggested that CA has a significant therapeutic benefit in the development of drug- induced acute liver failure by attenuating oxidative stress. A deep-going study on the biologic activity of CA will help in developing natural medicines-based therapeutic interventions for acute liver injury.

 XML Download
XML Download