

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Eosinophils are produced in the bone marrow along with other white blood cells and circulate at relatively low levels in the bloodstream, making up 1-3% of white blood cells. Eosinophils also occur outside the bone marrow and blood vessels: in the medulla and the junction between the cortex and medulla of the thymus and in the lower gastrointestinal tract, ovary, uterus, spleen, and lymph nodes. In allergic conditions, they are found in the lung, skin, and esophagus. Eosinophils persist in the circulation for 8-12 hours and can survive in tissues for an additional 8-12 days in the absence of stimulation.1 Eosinophils are distinguished based on their characteristic morphological features, namely bilobed nuclei and cytoplasmic granules of a distinctive granular pink,2 and they are about 12-17 µm in diameter (Fig. 1). While no cell surface proteins unique to eosinophils have as yet been recognized, they are armed with abundant specific cytoplasmic granules with their structural packaging of cationic proteins, the eosinophils' most characteristic morphologic feature. KoreaEosinophil granules are composed mainly of cytotoxic cationic proteins and also harbor a multitude of cytokines and chemokines. Eosinophils are terminally differentiated cells that arise from hematopoietic CD34+ stem cells through commitment and differentiation and do not appear to multiply after leaving the bone marrow. An interplay of several key transcription factors dictates eosinophil lineage development and differentiation, and an almost identical set of factors activates transcription of eosinophil-specific genes encoding the major basic protein (MBP), eosinophil-derived neurotoxin (EDN), eosinophil peroxidase (EPO), Charcot-Leyden crystal (CLC) protein, CC chemokine receptor 3 (CCR3), and interleukin-5 receptor alpha (IL-5Rα) chain. Eosinophils are multifunctional leukocytes implicated in the pathogenesis of inflammatory responses, notably including allergic diseases and parasitic helminth infections. Much controversy exists as to the role of eosinophils in homeostatic and diseased conditions. Recent advances have allowed selective removal of eosinophils in rodents and asthmatic patients through genetic manipulation and therapeutic agents. With these tools, we are now in a much better position to determine the role of eosinophils in the pathophysiology of asthma and so develop novel therapeutic approaches.
Eosinophil development
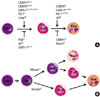
Recent advances in the biology of cellular development/differentiation have highlighted the fact that any cell type can seemingly become any other, given the correct combinations of transcription factors and environment. Eosinophil development appears to faithfully conform to this notion. Eosinophil lineage fate is determined by the interplay of a few key transcription factors, including CCAAT/enhancer-binding protein (C/EBP family member), GATA-1 (a zinc finger family member), PU.1 (an Ets family member), and friend of GATA (FOG). In particular, C/EBPα/β and GATA-1, either individually or in concert, play a decisive role in eosinophil commitment from multipotent stem cells. C/EBPs are a family of transcription factors that contain a highly conserved, basic-leucine zipper domain at the C-terminus that is involved in dimerization and DNA binding. Six members of this family (α, β, γ, δ, ε, and ζ) have thus far been isolated and characterized.3 Expression of NF-M, the chicken homolog of C/EBPβ, fused to the ligand binding domain of estrogen receptor induces the up-regulation of an eosinophil-specific surface marker EOS47 along with the down-regulation of a specific marker of a multipotent chicken progenitor cell line transformed by the Myb-Ets oncoprotein.4 Mice with a null mutation in C/EBPα fail to generate eosinophils and neutrophils, whereas other hematopoietic lineages, including monocytes, are not affected.5 The enforced expression of either C/EBPα or C/EBPβ induces eosinophil differentiation of the chicken-transformed cells.6 A dominant negative C/EBP that antagonizes all C/EBP members blocks granulocyte and monocyte development from human cord blood CD34+ progenitors.7 There is a functional redundancy of C/EBPα and C/EBPβ family members for granulocyte development/differentiation, although C/EBPβ-deficient mice do not show any defects in formation of myeloid lineage, unlike C/EBPα.8 Additionally, a dominant negative C/EBPβ phenotype induces the formation of immature eosinophils, indicating that C/EBPβ also promotes eosinophil maturation.6
GATA-1 is a member of the GATA family of transcription factors that contain two zinc finger motifs. GATA-1 is expressed in the hematopoietic system, including by erythroid cells, megakaryocytes, eosinophils, and mast cells and in the Sertoli cells of the testis.9 GATA-1 reprograms avian myeloblastic cell lines to eosinophils, and its expression level fine tunes development of the eosinophil lineage.10 Human cord blood CD34+ cells that are transduced by GATA-1-expressing retrovirus exclusively give rise to eosinophils. The C-terminal zinc finger motif of GATA-1 is necessary for formation of eosinophils, and GATA-1-deficient fetal liver cells lack the ability to form eosinophils.11 Deletion of a high-affinity GATA-binding site in the GATA-1 promoter, ΔdblGATA, leads to selective loss of the eosinophil lineage, whereas development of platelet, mast cells, and red blood cells remains little-changed.12 When granulocyte/macrophage progenitors (GMPs) are isolated from bone marrow cells of transgenic GATA-1 reporter-tagged GFP and grown in liquid culture, eosinophils are found only in the GFP-positive fraction, along with acquisition of surface IL-5Rα.13 C/EBPβ and GATA-1 synergistically regulate activity of MBP promoter.14 The level of GATA-1 expression is an important element in establishing the eosinophil phenotype, as it activates an eosinophil-specific gene at low, and represses it at high, GATA-1 concentrations.10,15 Additionally, the timing of expression of these transcription factors is critical. For instance, when GATA-2 acts on GMPs expressing C/EBPα, it exclusively induces eosinophil formation, whereas it instructs GMPs to form basophils and/or mast cells if GMPs are not expressing C/EBPα.16 GATA-2 has an instructive capacity toward eosinophil lineage from human cord blood CD34+ progenitors comparable to that of GATA-1 and efficiently compensates for GATA-1-deficiency in terms of eosinophil development in vivo.11 GATA-2 also complements GATA-1 to activate EDN transcription.17 Nonetheless, the in vivo role of GATA-2 in eosinophil development remains to be determined, as GATA-2-deficient mice display a general reduction in hematopoiesis, and a complete lack of mast cells.18
Given that the two transcription factors GATA-1 and C/EBP serve as the master regulators of eosinophil development, it is proper to mention how GATA-1 and C/EBP might induce eosinophil formation in CD34+ cells. Two models have been proposed.19 In the first, stochastic expression of either GATA-1 or C/EBPα in a common progenitor induces expression of the other, resulting in co-expression of both factors and ultimately eosinophil formation. In the second model, each of the factors acts on a distinct type of CD34+ cell, leading to production of eosinophil lineage. These authors favor the second model, as several distinct subpopulations of CD34+ cells exist, and Myb-Ets-transformed multipotent progenitor cells can readily be converted to any cell type depending on the combination of transcription factors, including C/EBP, GATA-1, PU.1, and FOG (see below), to which they are exposed.
Other players also act in concert with C/EBP and GATA-1 in the process of eosinophil commitment. PU.1 is a transcription factor with a winged helix-turn-helix DNA binding domain that is a member of the Ets transcription family and is expressed in hematopoietic cells, including myeloid cells.20 Conditional activation of PU.1 in Myb-Ets-transformed multipotent progenitor cells induces the formation of cells with properties of immature eosinophils after short-term culture.21 The mechanism by which PU.1 induces eosinophil commitment in transformed cells involves downregulation of GATA-1 expression,22 agreeing with the observation that an intermediate GATA-1 level is required for eosinophil commitment.10,15 When PU.1 is co-expressed with C/EBPε32 and GATA-1, however, it transactivates the MBP promoter.22 Hence, PU.1 differentially exerts its function depending on the context of available transcription factors. FOG contains nine zinc fingers, at least two of which are capable of binding to the N-terminal finger motif of GATA-1.23 Expression of FOG in eosinophils leads to a loss of eosinophil markers and the acquisition of a multipotent lineage, and constitutive expression of FOG in multipotent progenitors inhibits activation of MBP gene transcription by GATA-1,14 C/EBPβ,24 or a combination of GATA-1, C/EBPε32, and PU.1.22 Thus, FOG acts as a repressor of the eosinophil lineage. These results highlight the importance of both cooperative and antagonistic interactions of multiple transcription factors for eosinophil-lineage commitment from multipotent hematopoietic progenitors.
The involvement of two additional transcription factors in eosinophil development has been documented. IFN consensus sequence binding protein (Icsbp) is an IFN-γ-induced transcription factor that regulates IFN-responsive genes.25 Icsbp-deficient mice have reduced eosinophil developmental potential and eosinophil progenitors. Eosinophil progenitors from icsbp-deficient mice show reduced expression of GATA-1 and are unable to respond to IL-5 in terms of eosinophil colony formation.26 Therefore, Icsbp appears to play a critical role in the development of the eosinophil lineage, although little known about the underlying molecular mechanism. Id proteins are basic helix-loop-helix transcription factors that lack a basic DNA binding domain.27 Constitutive expression of Id1 inhibits eosinophil development, whereas Id2 accelerates the final maturation of eosinophils. The effects of Id factors do not seem to be restricted to eosinophils, however, because they also promote neutrophil development and maturation.28 Notch is an evolutionarily conserved transmembrane protein that regulates a broad spectrum of cell-fate decisions and differentiation.29 Notch signaling promotes eosinophil maturation30 as well as affecting eosinophil functionality.31,32 However, whether Notch signaling modulates the transcription factors or unidentified pathways key for deciding eosinophil fate or is itself affected by these factors remains to be determined.
Once multipotent progenitor cells commit to becoming eosinophil progenitors, they go through several intermediate stages before becoming fully mature eosinophils that are seen in the circulation and tissues. These stages include promyeloblasts, promyelocytes, metamyelocytes, band form, and segmented form, based on morphological criteria.33 As readouts in most studies of hematopietic development are measured by the formation of fully mature cells, it is difficult to identify the exact development/differentiation stage at which a particular transcription factor exerts its function. Most of the aforementioned transcription factors positively or negatively act on the commitment stage. These include GATA-1, GATA-2, C/EBPα, C/EBPβ, C/EBPε, PU.1, Icsbp, FOG, and Id1, whereas fewer numbers of transcription factors are known to specifically act on the late or terminal stages (Fig. 2A). For instance, Id228 and moderate GATA-1 levels10,15 are required for progression through maturation, whereas C/EBPε14/27 isoforms, which are highly expressed on peripheral blood eosinophils34 and terminally differentiated eosinophils,35 block MBP transcription. Given that a wide spectrum of transcription factors is present in mature eosinophils, they may influence late-stage eosinophil differentiation and maturation. These molecules could be novel targets for therapeutic approaches to eosinophil-associated inflammation.
Despite sharing many features, such as transcription factors for eosinophil commitment and maturation, there are subtle differences in the lineage pathway through which eosinophils are generated in mice and humans (Fig. 2B). In mouse hematopoiesis, eosinophil potential exists along with the granulocyte/monocyte differentiation pathway from hematopoietic stem cells, and at least a fraction of granulocyte/macrophage progenitors (GMPs) are bipotent for the eosinophil and the neutrophil lineages. Eosinophil progenitors are found within cells activating GATA-1, whereas GMPs that do not express GATA-1 give rise to neutrophils and macrophages. Thus, eosinophil progenitors exist as a distinct population downstream of GMP.13 The mouse bipotent basophil/mast cell progenitor and the basophil lineage-committed progenitor are also identified downstream of the granulocyte/macrophage progenitor,36 suggesting that the commitment of eosinophil and basophil/mast lineages occurs independently after the multipotent progenitor has lost the megakaryocyte/erythroid lineage potential. In contrast, in human hematopoiesis, GMPs lack eosinophil potential, and eosinophil progenitors are instead found in common myeloid progenitors (CMPs) that both do and do not express surface IL-5Rα. Cells expressing IL-5Rα give rise exclusively to eosinophils but never basophils or neutrophils.37 However, as cells possessing both basophil and eosinophil granules have been found in leukemia patients,38 it is possible that a distinct cell type exists that has deviated from the known lineage pathway.
Regulation of eosinophil-specific genes
Analysis of the transcription factors that control eosinophil-specific genes may offer insights, at the molecular level, into the mechanisms behind the commitment of multipotent progenitors into the eosinophil lineage. Relatively small numbers of genes are exclusively or predominantly transcribed in eosinophil progenitors and fully differentiated eosinophils.39 These include genes encoding eosinophil granule proteins (MBP, ECP, EDN, EPO, CLC protein) and surface receptors (IL-5Rα and CCR3). Not surprisingly, almost the same set of transcription factors that dictate eosinophil commitment and differentiation are also involved in controlling transcription of eosinophil-specific genes. These include C/EBP family proteins, GATA factors, and PU.1. The regulatory regions of these genes include known or putative binding sites for these transcription factors. Interestingly enough, these cis-acting control elements are clustered in the sequences flanking their exon 1 and intron 1 rather than the promoter (Fig. 3), although the implication of this for transcriptional regulation remains to be determined.
MBP is a granule protein localized in the crystalline core with no known enzymatic activity. Transcriptional regulation of the human MBP gene is the most thoroughly studied of all eosinophil-specific genes14,22,34,40,41 because MBP is a representative marker of eosinophils, and MBP transcript accounts for up to 8.1% of the total cellular mRNAs of eosinophils.42 Two different transcripts arise from differential splicing of alternative MBP transcripts from promoters P1 and P2, respectively, located 32 kb apart in the genomic DNA. The P2 promoter is predominantly responsible for MBP expression in eosinophil lineage cells.40 The P2 promoter of the MBP gene contains a functional GATA site and a C/EBP site (Fig. 3A). Binding of GATA-1 or C/EBPα/β to its respective binding site transactivates the MBP P2 promoter.41 A subsequent study by the same group showed that GATA-1 and C/EBPβ interact physically to synergistically transactivate the MBP P2 promoter. Furthermore, FOG acts as a negative cofactor for GATA-1-, but not C/EBPβ-, mediated transactivation.14 The P2 promoter is activated by GATA-1 alone but is synergistically transactivated by low levels of PU.1 in the presence of optimal GATA-1 levels. PU.1 and C/EBPε individually activate the P2 promoter.34 In addition to GATA-1 and C/EBPβ, the combination of GATA-1 and PU.1 transactivates the MBP P2 promoter.34 By contrast, C/EBPε14, which lacks the transactivation domain and is expressed at high levels in terminally differentiated eosinophils,34,35 strongly inhibits the P2 promoter. C/EBPε27 also represses P2 promoter activity via protein-protein interaction through the C/EBP and/or GATA-binding sites, but not the PU.1 sites.34 Thus, the active transcription complex consisting of well-known transcription factors is required for regulation of MBP P2 promoter activity. The complex includes interactions between GATA-1 and C/EBPα, GATA-1 and C/EBPβ, GATA-1 and C/EBPε isoforms, GATA-1 and PU.1, PU.1 and C/EBPε isoforms, and GATA-1 and FOG.14,22,34,41 These findings establish a combinatorial cooperation and antagonism through protein-protein interactions of the transcription factors that control eosinophil development.
EDN is a cationic granule protein synthesized in eosinophils,43 and it has ribonuclease activity that can degrade the RNA genomes of some viruses. EDN also has an immunomodulatory function in terms of regulation of dendritic cell migration44. As seen in Fig. 3, the key regulatory sequence of EDN transcription resides in the promoter and intron, which contain GATA, C/EBP, PU.1, NFAT, and AP-1 sites,17,45 most of which are functional. PU.1,46 C/EBP isoforms α, β, and ε47, or NFAT binding48 to their respective binding sites in intron 1 of the gene induces transactivation. The promoter region also contains two GATA sites, which are 600 bp apart. GATA-1 and GATA-2 bind the two functional GATA sites in the EDN promoter. GATA-2 can replace the effect of GATA-1.17 Elsewhere, HNF4 interacts with Sp1 to stimulate EDN promoter activity.49
ECP is found in the matrix of the eosinophil-specific granule and has more potent anti-helminthic activity but less ribonuclease activity than EDN.50 The ECP gene sequence is highly homologous to that of EDN, in particular, with 92% identity in the upstream 1-kb sequence.51 Given that the ECP gene shares with the EDN gene all cis-acting elements at identical positions (Fig. 3), almost identical molecular cues appear to govern regulation of gene expression.
EPO is a heme-containing glycoprotein that possesses peroxidase activity. It is located in the matrix of the granule and has a sequence that is closely related to neutrophil myeloperoxidase.52 A number of positively and negatively cis-acting elements are mapped to the proximal promoter of this gene, including transcription factors Egr-1, H4TF-1, CTCF, UBP-1, and GaEII, although it is not known whether these potential binding sites are functional for EPO transcription.53 Additionally, sequence analysis shows that binding sites for GATA factors, PU.1, and C/EBP are present in intron 1 and the promoter (Fig. 3), again suggesting that the transcription factors for eosinophil development are actively involved in the transcriptional regulation of the EPO gene.
CCR3 is constitutively expressed at high levels in eosinophils, with 16,000-60,000 receptors per cell; it serves as the primary chemokine receptor responsible for eosinophil trafficking to tissues in diseased and healthy conditions.54,55 CCR3 is also expressed on prominent allergic inflammatory cells, including Th2 helper56 and mast cells.57 The restricted expression of CCR3 leads to a notion that it plays an integral role in the pathogenesis of allergic diseases including asthma, allergic dermatitis, and allergic rhinitis. Furthermore, as airway epithelial cells express functional CCR3, this protein is postulated to play roles beyond simple cell trafficking, such as in airway remodeling.58 A recent study revealed that CCR3 serves as an identification marker, along with IL-5Rα, in eosinophil progenitors at the very early stage of human eosinophil development.37 Hence, analysis of the transcription factors that control CCR3 expression may offer insights into the mechanisms behind the commitment of common myeloid progenitors to the eosinophil lineage. The key sequences for CCR3 gene transcription reside in exon 1 and intron 1 rather than in the promoter,59-61 (Fig. 2B). Multiple GATA binding sites are present in exon 1 and intron 1. Exon 1, in particular, has five GATA sites, each of which has a different GATA-1 binding affinity, with one of the five as a positively acting element and two as negatively acting elements for transcription in vitro.62 C/EBP and PU.1 binding sites are located in the promoter and intron 1 regions, respectively, although their function remains to be determined. Additionally, AML-1 and CREB binding motifs are present in exon 1. We recently found evidence that protein binding to AML-1 and CREB sites contributes to transactivation of the CCR3 gene (our unpublished results), as much as does GATA binding. Therefore, it is plausible that these transcription factors participate in eosinophil development and maturation.
IL-5R consists of heterodimer, a unique ligand-binding α chain and β chain shared with IL-3 and GM-CSF receptors that is linked to the Janus kinase/signal transducer and activator of transcription and phosphoinositol-3-kinase.63 IL-5R mediates their differentiation and maturation, survival, chemotaxis, and effector functions.64 Eosinophils, but not basophils or neutrophils, possess a high level of IL-5Rα, and IL-5Rα is a key surface molecule in sorting of murine eosinophil progenitors.13 Although IL-5Rα is expressed as a result of commitment to the eosinophilic lineage,13 human CMPs that express IL-5Rα give rise to only eosinophils.37 Therefore, IL-5Rα is presumably the earliest phenotypic marker that eosinophils acquire at the commitment step of the developmental pathway. Given that IL-5Rα-positive CMPs are derived from the CMPs that lack this surface marker, the signals and transcription factors that induce IL-5Rα transcription may play an integral role in eosinophil fate decision. An early study demonstrated that 34 bp of the proximal region of the IL-5Rα promoter serves as the binding site for a myeloid- and eosinophil-specific transcription factor.65 These turned out to be RFX family transcription factors, although RFX family proteins are not expressed in a myeloid- or eosinophil-specific manner.66 An AP-1 site, located upstream of the RFX binding site, functionally cooperates with a neighboring EOS site to mediate IL-5Rα transcription. C-Jun, CREB, and CREM bind to the AP-1 site.67 There is a second promoter, designated P2, for the human IL-5Rα gene. Oct2 transactivates murine B cells' IL-5α gene by binding to its promoter.68 A short sequence of 6 bp within the P2 promoter is responsible for the binding of an uncharacterized protein and is sufficient for promoter activity in eosinophilic cells.69 In C/EBPα-null mice, expression of the IL-5Rα gene was greatly reduced.69 Moreover, sequence analysis of exon 1 and intron 1 as well as promoters shows that a number of GATA factors, C/EBP, and PU.1 binding sites are concentrated in these sequences (Fig. 2B). Nevertheless, the important transcription factors GATA-1 and C/EBP, which are believed to direct cells toward the eosinophil lineage, have not yet been reported as necessary and/or sufficient for transcription of the IL-5Rα gene.
Although the aforementioned transcription factors are primarily responsible for the regulation of eosinophil-specific gene expression, their mere presence even in combination is not sufficient for induction of gene transcription. Many eosinophil-specific genes encoding eosinophil basic proteins, CCR3, and IL-5R are induced by modifiers of histone structure such as histone acetyltransferase inhibitors,14,46,53,60,65 and expression of many asthma-related inflammatory genes is affected by these agents.70 DNA methyltransferase inhibitors also have a high propensity to alter eosinophil-specific gene expression. Moreover, regulation of these gene products by microRNAs has not yet been reported. As the relative importance of role of epigenetic regulation has increasingly become evident, the study of epigenetic regulation of eosinophil-specific genes is vital. Taken together, these findings show that the deciphering of eosinophil-specific gene expression will provide both a molecular basis for eosinophil development and targets for novel therapies for the treatment of eosinophil-associated diseases.
Role of eosinophils in asthma
Eosinophils are associated with the pathogenesis of asthma, and the presence of eosinophils in the airway lumen and lung tissues is often regarded as a defining feature of this disease.71 The role of eosinophils in the pathogenesis of asthma is due to their ability to mediate terminal effector functions and innate immune responses by secreting a wide variety of cationic proteins, lipid mediators, and cytokines/chemokines. Furthermore, eosinophils are capable of bridging innate and adaptive immune responses by elaborating T cells, dendritic cells, and mast cells. The recent availability of genetically modified mice makes possible the elucidation of a causal relationship between eosinophil recruitment and the onset or progression of pulmonary pathologies associated with asthma and provides new insight into the role of eosinophils in the pathogenesis of the allergic disease. In these animals, eosinophils are depleted or overproduced by manipulating expression of transcription factors regulating eosinophil development, production of IL-5 and eotaxins, and expression of receptors responding to these cytokines, through transgenic systems, gene disruption, and neutralizing antibodies. Thus, much information on the role of eosinophils roles has been accumulated using experimental models. This section describes the contribution of eosinophils to the pathogenesis of allergic disease within the context of asthma.
Eosinophil-deficient mice
Two strains of mice that lack eosinophils were engineered in different genetic backgrounds. Removal of a high-affinity double GATA site from the GATA-1 promoter (Δdbl-GATA) in a BALB/c background selectively ablates eosinophils.12 When Δdbl-GATA mice are subjected to a standard experimental asthma protocol of sensitization and challenge with allergen, the absence of eosinophils does not protect the mice from AHR development, but are required for airway remodeling.72 However, Δdbl-GATA mice created in a C57BL/6 background show decreased allergen-induced AHR, T cell recruitment to the lung, and production of Th2 cytokines and chemokines (Table 1). Furthermore, adoptive transfer of eosinophils or CCL11/eotaxin-1 delivery to Δdbl-GATA BABL/c mice results in recruitment of lung T cells and restoration of airway inflammation.73 A second line of mice devoid of eosinophils, PHIL mice, was created in the C57BL/6 background by transgenic expression of diphtheria toxin A driven by the EPO promoter.74 In this line of mice, eosinophils are nearly completely deficient in all organs in which they occur under homeostatic conditions. Allergen challenge of these mice does not induce AHR or pulmonary mucus accumulation, suggesting a link between eosinophils and allergic pulmonary pathologies. The combined transfer of Th2-polarized OVA-specific transgenic T cells and eosinophils to PHIL mice, but not transgenic T cells alone, results in accumulation of the effector T cells and airway Th2 responses, suggesting that the primary role of pulmonary eosinophils is to elicit localized recruitment of effector T cells.75 These data support the central hypothesis that eosinophils are required for the recruitment of T cells to the lung and thus are not only terminal effector cells but also important modulators of allergic asthma.
IL-5- or IL-5Rα-deficient mice
IL-5 plays a role in the pathogenesis of eosinophilic inflammation and asthma. Airway allergen challenge in asthmatics induces expression of IL-5 by T cells,76 whereas increased levels of IL-5 and MBP can be detected in the airway of symptomatic asthmatics.77 IL-5-deficient mice in a C57BL/6 background fail to develop AHR and airway eosinophilia upon aeroallergen challenge (Table 2), suggesting an essential role for IL-5 in induction of eosinophilia and development of AHR.78 Indeed, reduced lung eosinophils and AHR are observed in mice treated with IL-5 antibodies.79 IL-5-deficient mice also show lesser alterations in tissue remodeling events, including peribronchial fibrosis and thickness of the peribronchial muscle layer, along with a reduction in the production of TGF-β and MBP by eosinophils.80 In contrast, IL-5-deficient BABL/c mice develop allergen-induced AHR, as wild-type mice do, despite markedly reduced blood and lung eosinophilia,81,82 suggesting dissociation of airway eosinophilia from AHR development. On the other hand, transgenic mice that constitutively express IL-5 in the lung epithelium develop an accumulation of eosinophils and pathologic changes including goblet cell hyperplasia, epithelial hypertrophy, and AHR even in the absence of antigen challenge.83 Genetic IL-5Rα deficiency decreases antigen-induced airway eosinophilia and AHR.84 The confusion involving the effect of IL-5 on lung functions is also observed in human clinical studies. An initial study using humanized anti-IL-5 antibody in patients with mild asthma demonstrated >90% lower blood and sputum eosinophilia but was not effective in improving lung function, as measured by FEV1.85 A subsequent study showed that anti-IL-5 did not reduce the level of MBP in the airways, even in the presence of partially inhibited airway eosinophils (approximately 55%).86 In contrast, anti-IL-5 therapy was effective in treatment of a small group of patients with eosinophilic asthma.87,88 Thus, studies from both human subjects and murine models show that IL-5 is responsible for the induction of pulmonary eosinophilia, but the role of IL-5-induced eosinophils in the pathogenesis of asthma remains unanswered. Nevertheless, the association/dissociation of airway eosinophilia with lung function seen in some mouse strains and the differential clinical benefits of anti-IL-5 therapy have important implications for the treatment of asthma and testify to the complex pathogenesis of the disease.
Eotaxins (CCL11, CCL24, and CCL26)- and/or CCR3-deficient mice
Three eotaxin family proteins, eotaxin-1/CCL11, eotaxin-2/CCL24, and eotaxin-3/CCL26 have been identified,89 all of which selectively bind to CCR3. Eotaxin-2 and -3 are distantly related to eotaxin-1, with ~30% sequence identity and different chromosomal locations. Gene disruption studies of eotaxins-1 and -2 have been published, and both eotaxin-1 and eotaxin-2 have not yet been characterized as a functional murine homologue of eotaxin-3 (Table 2). Targeted disruption of CCL11/eotaxin 1 leads to partially reduced eosinophil counts in the blood and airways under baseline conditions without affecting eosinophil hematopoiesis in the bone marrow. Upon exposure to aeroallergen, eotaxin-1-deficient mice show ~70% reductions in eosinophil numbers in the airway compared with un-sensitized wild-type mice,90 but they retain substantial levels of pulmonary eosinophils. The same knock-out mice have a selective reduction (approximately 95%) in eosinophil counts in the jejunum and thymus, indicating that eotaxin-1 is a fundamental regulator of the physiological trafficking of eosinophils in the body during health.91 However, eotaxin 1-deficient mice, whose eotaxin gene had been replaced with a transgenic Escherichia coli β-galactosidase gene, developed lung eosinophilia in response to allergen challenge and had no histologic or hematologic abnormalities,92 contradicting two earlier studies.90,91 Another study suggested that eotaxin 1-deficient mice in a BALB/c background possessed no defect in the development of allergen-induced AHR and blood eosinophila, with partially reduced airway eosinophilia,82 suggesting that incomplete elimination of lung eosinophils is not sufficient to abolish AHR. Eotaxin-2-deficient mice have normal baseline eosinophil levels in the hematopoietic tissues and gastrointestinal tract. However, these mice do not develop airway eosinophilia in response to IL-13. Additionally, IL-13 induces eotaxin-2, but not eotaxin-1, expression by macrophages in BALF. These results suggest non-redundant roles for these two CCR3 ligands in response to inflammatory airway environments.93 Eotaxin-1/2 double-deficient mice exhibit a profound decrease in eosinophils in BALF and peribronchial tissue compared with mice carrying a single deletion, comparable to the effect in CCR3-deficient mice.94 Additionally, eotaxin-1 and eotaxin-2 contribute to lung pathology differently: eotaxin-1 is important in the development and maintenance of peribronchial eosinophilia,95 whereas eotaxin-2 is primarily responsible for IL-13-induced airway epsonophilia.93 Another type of double knock-out BALB/c mice that are deficient in both IL-5 and eotaxin-1 fail to develop allergen-induced AHR and completely lack eosinophils in the blood and lungs, whereas either IL-5 or eotaxin-1-deficient mice develop AHR, as do wild-type mice, suggesting that complete removal of airway eosinophils is required to impede AHR development. Additionally, Th2 cells in these mice produce reduced IL-13 levels, a critical regulator of pathologic changes in the asthmatic lung, indicating that eosinophils can link to adaptive immune responses by modulating CD4+ T cell functions.82
Analysis of CCR3-deficient mice shows that a lack of CCR3 results in markedly reduced eosinophil recruitment to the lung, with the majority of eosinophils trapped in the subendothelial space. However, CCR3-deficient mice unexpectedly exhibit greater airway responses to methacholine than do wild-type mice when subjected to systemic sensitization followed by respiratory antigen challenge, indicating that CCR3 disruption confers no protection, but rather exacerbates AHR.96 However, allergen-challenged CCR3-deficient mice fail to develop AHR upon epicutaneous sensitization.97 Therefore, it is not clear whether CCR3 is the dominant pathway in chronic models of allergic airway inflammation. Moreover, CCR3-deficient mice have more mast cells in the airways after antigen challenge,96 reflecting a more complex role for CCR3 in the pathological events of asthma. In contrast to the conflicting findings from CCR3-deficient mice, administration of anti-CCR3 antibody via both systemic and local routes abolishes antigen-induced lung eosinophilia and AHR.98
To summarize the gene-ablation studies, two axes, IL-5/IL-5R and eotaxins/CCR3, play dominant roles in allergen-induced pulmonary eosinophilia. However, the contribution of eosinophils to the pathogenesis of this allergic disease has been controversial, depending on the rodent strain (largely C57BL/6 vs. BALB/c mice), experimental protocol (e.g. aerosol versus cutaneous routes, chronic cytokine exposure versus allergen challenges, and severity of antigen challenge), and pathological conditions (e.g. the milieu of Th2 cytokines present in the lung). Moreover, conflicting effects of eosinophil depletion are observed in human diseases. Nevertheless, the discrepancies in the pathological phenotypes reflect the heterogeneous nature of asthma in humans and have important implications for selection of therapeutic targets and designing therapeutic agents.
CONCLUSIONS
Understanding of eosinophil development, trafficking, and effector function may lead to the development of a core experimental instrument, reduction or elimination of eosinophils in asthma model and human subjects. The anti-eosinophil approaches allow intense testing of the link of eosinophils to the lung functions and pathologies of asthmatic lungs, prove useful to identify critical pathways involved in the recruitment and activation of eosinophils in the asthmatic lung, and draw attention to the potential of anti-eosinophil-directed therapeutics. Despite increasing knowledge in eosinophil's role by the use of eosinophil-deficient mice in models of disease, none of these models fully reflects the human disease. Furthermore, these models might not be predictive of the role played by the eosinophil in the human disease. This is at least in part due to the fact that the causative relationship between eosinophil activities and the onset/progression of allergic respiratory pathology is affected by a variety of pathologic conditions and inflammatory microenvironments in the lung and system. Further studies are needed to clarify role of eosinophils in diverse disease settings and to identify the downstream mechanism, such as cooperation with resident lung cells. Such analyses will help to establish pathophysiological paradigms and to uncover the molecular insight into disease pathogenesis.



 XML Download
XML Download