

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Replicative senescence is an irreversible growth arrest of primary cells in culture system. It has been reported that aging in vivo is related to the limited replicative capacity that normal somatic cells show in vitro (Hayflick & Moorhea, 1961). Telomere shortening theory has been linked to replicative senescence (Campisi, 2001). Absence of telomerase in somatic cells causes the progressive shortening of telomeric DNA. This erosion eventually destroys telomere integrity and triggered senescence by activating DNA damage checkpoints that block further cell division (Smogorzewska & de Lange, 2002). However, it has been reported that many stimuli, including DNA damage and oxidative stress, result in cells to arrest growth with a senescent phenotype, independent of telomere length (Fagagna et al., 2003; Gorenne et al., 2006; Petersen et al., 1998; Sharma et al., 2006). Since other than human primary somatic cells like mouse and dog fibroblast cells are known to contain telomerase activity and longer telomeres, telomere shortening theory can not apply to other mammalian somatic cells.
Increasing number of evidence, mostly from work in fibroblasts, indicates that oxidative stress can also induce replicative senescence (Serrano & Blasco, 2001). Reactive oxygen species (ROS) produced during oxidative phosphorylation in mitochondria attack cellular macromolecules and produce oxidized protein, lipid and DNA products. Damaged macromolecules are accumulated with age in animal cells (Hamilton et al., 2001; Levine et al., 2002; Pamplona et al., 2000). High levels of oxidized cellular macromolecules have been reported to be correlated with shorter life spans in mammals (Barja & Herrero, 2000). Cultivation of fibroblast in lower oxygen, instead of atmospheric oxygen resulted in delaying of replicative senescence, indicating that oxidative stress contributes to replicative senescence (Parrinello et al., 2003).
Ascorbic acid is an antioxidant that can protect cells from oxidative stress. When ascorbic acid was treated in hydrogen peroxide-treated human dermal fibroblast cells, ascorbic acid protected the hydrogen peroxide-driven oxidative damage. Ascorbic acid has also been reported to prevent damage of cellular macromolecules including lipid, protein, and DNA (Amer, 2002; Carty et al., 2000; Huang et al., 2002; Lenton et al., 1999) and incidence of certain cancers (Chen et al., 1988). However, life-long ascorbic acid supplementation has shown controversial effects in animal studies. A life-long supplementation of ascorbic acid or mixed antioxidants in the drinking water of mice was reported to increase mean life span significantly (Massie et al., 1984; Veurink et al., 2003). But a mixed antioxidant diet had no effect on life span of rats (Holloszy, 1998; Selman et al., 2006). Ascorbic acid also resulted in the extension of replicative life span in human diploid fibroblast culture. The extension of life span was accompanied by reducing the rate of telomere shortening (Farriol et al., 1994; Kashino et al., 2003; Levine, 2002). However, the detailed molecular mechanism other than telomere shortening has not been investigated. In this paper, we attempted to elucidate how ascorbic acid extends replicative life span of HEF cells. Here, we showed that ascorbic acid postponed replicative senescence of human fibroblast by reducing mitochondrial and DNA damages through scavenging ROS.
Materials and Methods
Cell culture and cell cycle analysis
A primary human embryonic fibroblast (HEF) that was established from an abortus at Hallym University Hospital was obtained (Kim et al., 2005) and used for this work. Human embryonic fibroblasts were grown in DMEM supplemented with 10% FBS at 37℃ with humidified atmosphere containing 5% CO2. The primary culture was designated as zero of PD number. HEF cells were cultivated continuously until replicative senescence by splitting cells (at 1:4 splitting rate) every 3-6 days and the media were replaced every 3-4 days. HEF cells were treated continuously with various concentrations of ascorbic acid from PD 40 and measured the maximum PD number to test the extension of PD. Since it was reported that blood Vitamin C level maintained at 24 µM and oral supplementation of Vitamin C increased blood Vitamin C level, ascorbic acid was treated at 2, 20, and 200 µM (Fumeron et al., 2005). Plating efficiency was analyzed by counting the number of floating cells 1 day after splitting to calculate correct PD numbers. For cell cycle analyses, 2 × 106 cells were harvested by trypsinization and fixed by cold 70% ethanol. After fixation at -20℃ for 4 hr, cells were collected and subsequently resuspended in staining solution (50 µg/mL propidium iodide, 50 µg/mL RNase A), and subjected to FACScan analysis using a FACS Vantage flow cytometer. Experiments were repeated more than three times to confirm the result.
SA-β-gal staining
Senescence associated-β-galactosidase (SA-β-gal) activity was determined by the procedure described previously (Dimri et al., 1995). HEF cells were washed with 1 × PBS and then fixed with 2% formaldehyde in PBS for 5 min. Cells were washed again two times with PBS and then mixed with staining solution (1 mg/mL X-gal, 40 mM citric acid/sodium phosphate (pH 6.0), 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl2) and incubated at 37℃ for 18 hr. After incubation, the percentage of stained cells was counted under the microscope. Average value and standard deviation calculated from more than three repeats of experiment were used.
Measurement of ROS level and age-dependent mitochondrial damage by fluorometer
The level of cellular ROS was measured through fluorometric detection of DCFH (2', 7'-dichlorodihydrofluorescin) oxidation induced by ROS. Age-dependent mitochondrial damage was measured through fluorometric detection of Rho123 (dihydrorhodamine 123). Cells were incubated with 10 µM DCFH at 37℃ for 10 min. They were then washed twice with PBS, and lysed with lysis buffer [10 mM Tris-HCl buffer (pH 7.4) containing 0.5% Tween 20]. Cell debris was removed by centrifugation at 10,000 × g for 10 min. DCF fluorescence of the supernatant was measured using a spectrofluorometer with excitation of 500 nm and emission by scanning from 500 to 550 nm. Accumulation of Rho123 in mitochondria was measured with excitation at 500 nm and emission at 536 nm. Average value and standard deviation calculated from more than three repeats of experiment were used.
Analysis of aconitase activity
Aconitase activity was determined as described (Liang et al., 1997). 4 × 106 cells were incubated in ice-cold reaction buffer [50 mM Tris (pH 7.4), 0.6 mM MnCl2, 20 mM fluorocitrate] containing a mixture of protease inhibitors (5 µg/mL leupeptin, 5 µg/mL aprotinin, and 1 mM PMSF) and disrupted by sonication with three bursts of 5 seconds on ice. After centrifugation, the supernatant was used for measuring total soluble protein. Aconitase activity was determined spectrophotometrically as described previously (Levine, 1997). Average value and standard deviation calculated from more than three repeats of experiment were used.
Western blot analysis
HEF cells were harvested, washed in PBS, and lysed with extraction buffer [50 mM Tris-Cl (pH 7.4), 150 mM NaCl, 5 mM EDTA (pH 8.0), 1% NP-40] containing a mixture of protease inhibitors (1 mM PMSF, 5 µg/mL aprotinin, 5 µg/mL leupeptin). After centrifugation, the protein concentration of the supernatants was determined using BCA protein assay reagent (Pierce). The proteins were loaded on a 10% or 12% SDS-PAGE gel and separated. The resulting gel was transferred onto PVDF filter, and the filter was blocked in 5% nonfat powdered milk in TBST [50 mM Tris (pH 7.5), 150 mM NaCl, 0.1% Tween 20]. The blots were probed with anti-p21 monoclonal antibody (Pharmigen), anti-p53 monoclonal antibody (DO-1, Santa Cruz), and antiphospho-p53 (ser-15, Santa Cruz). The bands were visualized with the ECL kit according to manufacturer's instruction (Amersham).
AP site assay
AP (apurinic/apyrimidinic) site assay was performed as previously described (Nakamura et al., 1998). HEF cells in 5 mM glucose/PBS were incubated with 3 mM ARP (aldehyde reactive probe) at 37℃ for 60 min. The resulting cells were then collected by centrifugation and washed twice with PBS. The genomic DNA was isolated by chloroform/phenol extraction. One microgram of DNA was dissolved in 200 µL of 10 mM Tris buffer (pH 9.0) and mixed with 15 µL of 5 M NaCl. The resulting mixture was incubated at room temperature for 60 min with 30 µL of avidin-HRP. The DNA-avidin-HRP complex (DNA-HRP) was separated from unbound avidin-HRP by gently mixing 65 µL of 1 mM DAPER {N,N'-bis [3,3' (dimethylamino) propylamine-3,4,9,10-perylenetetracarboxylic diimide]} and incubated at room temperature for 5 min. The DNA-DAPER precipitate was then collected by centrifugation at 4℃ at 12,500 × g for 5 min. The pellet was resuspended by pipetting into wash buffer [170 mM NaCl, 20 mM Tris (pH 8.0), 0.25% Tween 20, 1% BSA], followed by centrifugation at 4℃ at 12,500 × g for 5 min. DNA-HRP precipitate was suspended in ice-cold 50 mM Na-citrate (pH 5.3) and sonicated for 2-3 sec and cooled immediately. AP site formation was measured HRP activity, as an indicator of AP sites in DNA-HRP by using fluorogenic QuantaBlu substrate kits (Pierce). Average value and standard deviation calculated from more than three repeats of experiment were used.
Results
Ascorbic acid treatment extended replicative life span of HEF cells
In order to test whether ascorbic acid extends replicative life span of HEF, HEF cells were treated continuously with various concentrations of ascorbic acid from PD 40 and measured the maximum PD number. Cells were cultivated continuously in 100-mm dishes by changing fresh media containing ascorbic acid once in every three days. Ascorbic acid treatment (at 200 µM) extended maximum population doubling of HEF cells by about 18% (11 PD) (Fig. 1A). The treatment of 2 µM ascorbic acid extended maximum PD by 2-3.5 over the untreated control (Table 1). High concentration of ascorbic acid, (higher than 2 mM) turned out to be toxic to the cells and led cells to death (data not shown). To confirm the effect of ascorbic acid, we also tested SA-β-gal staining, an in vitro and in vivo aging marker, at PD 64. Senescent HEF cells have been known to show a marked increase of blue colored SA-β-gal staining in perinuclear area. As expected, HEF cells treated with ascorbic acid dramatically decreased SA-β-gal-positive staining compared to untreated-HEF cells or H2O2-treated HEF cells (Fig. 2A, B). Because hydrogen peroxide-treated HEF cells showed earlier replicative senescence than untreated HEF cells, we measured Rho123 at PD 58. HEF cells treated with 200 µM ascorbic acid lowered SA-β-gal positive staining by 2.3 folds staining and the treatment of cells with 2 µM decreased by about 20% in percentage of SA-β-gal-stained cells. H2O2-treated HEF cells showed increased SA-β-gal positive staining by about 10% as expected. The result clearly showed that ascorbic acid treatment extended replicative life span in HEF cells.
Ascorbic acid treatment lowered ROS level in HEF cells
Since reactive oxygen species (ROS) known to damage progressively vital macromolecules and thereby contribute to the processes of aging and cancer formation, we measured cellular ROS levels by estimating the oxidation of DCFH in HEF cells. HEF cells were treated continuously with ascorbic acid from PD 40. Cells were harvested at PD 64 and were used for ROS analysis. Cells treated with ascorbic acid exhibited much less ROS when compared with untreated HEF cells (Fig. 3A). HEF cells treated with 200, 20, and 2 µM ascorbic acid showed 5, 3, and 3 relative fluorescence of ROS formation but untreated HEF cells showed about 20 relative fluorescence of ROS formation. The result indicates ascorbic acid lowered ROS level by 6.7 folds. Because hydrogen peroxide-treated HEF cells showed earlier replicative senescence than untreated HEF cells, we measured ROS formation at PD 58. The treatment of ascorbic acid dramatically decreased ROS even in H2O2-treated HEF cells (20 µM) (Fig. 3B).
Ascorbic acid treatment prevents age-dependent decline of mitochondrial function in HEF cells
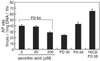
Since ROS is known to be generated in mitochondria and mitochondria are the major target of ROS, we attempted to measure mitochondrial damage by analyzing Rho123. The Rho123 fluorescence that accumulated in HEF cells was measured by fluorometer. HEF cells were treated continuously with ascorbic acid from PD 40. Cells were harvested at PD 64 and were used for Rho123 analysis. Senescent HEF cells showed high accumulation of Rho123 while young HEF cells showed much less levels of Rho123, indicating that senescent HEF cells have much higher mitochondrial damage. Rho123 accumulation was decreased by 70, 67, and 60% when treated with 200 µM, 20 µM, and 2 µM ascorbic acid, respectively (Fig. 4A). Because hydrogen peroxide-treated HEF cells showed earlier replicative senescence than untreated HEF cells, we measured Rho123 at PD 58. Hydrogen peroxide treatment exhibited much higher accumulation of Rho123 (by about 35%) as expected. Treatment of cells with ascorbic acid also lowered hydrogen peroxide-induced accumulation of Rho123 (Fig. 4B). Since age-dependent decline in aconitase activity as a marker of mitochondrial dysfunction was reported (Levine, 1997), we examined the effect of ascorbic acid on age-dependent decrease of aconitase activity. Ascorbic acid-treated HEF cells (at 200 µM) showed about 41% increases in aconitase activity when compared to untreated old HEF cells. These results suggest that ascorbic acid protects against age-dependent decline of mitochondrial function (Fig. 4C).
Ascorbic acid treatment relieved age dependent G1 arrest in HEF cells
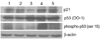
Since senescent cells have known to show G1 arrest, we tested whether ascorbic acid treatment lowers senescence-dependent G1 arrest. HEF cells were treated continuously with ascorbic acid from PD 40. Cells were harvested at PD 62 and were used for cell cycle analysis. Ascorbic acid treated-HEF cells (20 and 200 µM) showed considerable decrease from 64% to 52% in G1 population (Fig. 5). Further western blot analysis of cell cycle markers at HEF cells (old: PD 54, senescent: PD64) and ascorbic acid-treated HEF cells (old: PD 54, PD64, senescent: PD72) showed that ascorbic acid treatment markedly decreased the levels of p53, phospho-p53 at ser 15, and p21, indicating that ascorbic acid treatment relieved senescence-related G1 arrest (Fig. 6).
Ascorbic acid treatment decreased age dependent DNA damage in HEF cells
In order to examine whether ascorbic acid treatment reduces age dependent DNA damage, ascorbic acid-treated HEF cells were tested for AP site, a DNA base damage. HEF cells were treated continuously with ascorbic acid from PD 40. Cells were harvested at PD 64 and were used for AP site analysis. As expected, old HEF cells (PD 64) had about 68% higher AP sites compared to young HEF cells (PD 30). H2O2-treated HEF cells (PD 58) accumulated about 55% higher AP sites than untreated old HEF cells did. HEF cells treated with ascorbic acid decreased AP sites by 35% at 200 µM, by 13.7% at 20 µM, and by 9.8% at 2 µM when compared with untreated HEF cells (PD 64) (Fig. 7). The result suggested that ascorbic acid treatment markedly decreased age dependent DNA damage.
Discussion
Ascorbic acid has been reported to extend replicative life span in human diploid fibroblast culture. The extension of replicative life span was accompanied by reducing the rate of telomere shortening (Farriol et al., 1994; Kashino et al., 2003). However, the detailed molecular mechanism how ascorbic acid treatment extends replicative life span in human diploid fibroblast other than preventing telomere shortening has not been investigated. In this study, we attempted to elucidate molecular mechanism for the extension of replicative life span in HEF cells by ascorbic acid treatment. HEF cells were treated continuously with ascorbic acid from PD 40 untreated cells and harvested at PD 64 (senescent state in untreated HEF cells) for analyses of ROS, mitochondrial damage, G1 population, and DNA damage. ROS level estimated by analyzing DCFH was decreased dramatically by ascorbic acid treatment. Mitochondrial function assessed by Rho123 accumulation and aconitase activity was increased up to about 2 folds by ascorbic acid treatment (Fig. 4A, B). DNA damage measured by AP sites was decreased remarkably by ascorbic acid treatment (Fig. 7). G1 population examined by flow cytometry and western blot analysis of G1 markers (p21, p53, phospho-p53) was decreased markedly by ascorbic acid treatment (Fig. 5, 6) (Kim et al., 2005). Our result suggests that ascorbic acid extends replicative life span of HEF cells possibly by increasing growing population of HEF cells through lowering cellular ROS level and thereby reducing mitochondrial and DNA damages.
We also tested the effect of hydrogen peroxide treatment in addition to ascorbic acid treatment, as an additional oxidative stress. 20 µM of hydrogen peroxide was continuously treated beginning from PD 40 of HEF cells. H2O2 treatment resulted in apparently a premature senescence due to additional oxidative stress. H2O2-treated HEF cells decreased replicative life span by 6-8 PD compared to untreated HEF cells. Ascorbic acid treatment reversed this decrease induced by H2O2 treatment (Fig. 1). H2O2-treated HEF cells exhibited 13% higher SA-β-gal staining positive population as expected (Fig. 2). H2O2 treatment highly increased DCFH (60% increase) and Rho123 (40% increase) but ascorbic acid treatment reversed dramatically both DCFH and Rho123 to the level of H2O2-untreated HEF cells (Fig. 3, 4). AP sites were also increased greatly (55% increase) by H2O2 treatment (Fig. 7). Overall, H2O2 treatment speeded up the accumulation of oxidative stress by providing additional cellular oxidative stress and thereby accelerated replicative senescence, confirming that oxidative stress is a genuine contributor to establish replicative senescence.
Reduction in the rate of telomere shortening by ascorbic acid treatment may also contribute to the extension of replicative life span of HEF cells as reported previously (Farriol et al., 1994; Kashino et al., 2003). However, telomere shortening appears to be a part of DNA damage since telomere shortening activates DNA damage checkpoints that block further cell replication (Smogorzewska & de Lange, 2002). Ascorbic acid scavenges ROS and thereby seems to relieve random attack of telomere by ROS. This implies that telomere shortening may be a part of oxidative stress. In this context, it is understandable that many stimuli, including DNA damage and oxidative stress, cause cells to arrest growth with a senescent phenotype, independently of telomere length (Hart & Setlow, 1974; Parrinello et al., 2003). In addition, other mammalian primary somatic cells like mouse and dog fibroblast cells are known to contain considerable telomerase activity and longer telomeres. Mouse fibroblast cells still undergo replicative senescence after a limited number of population doublings when cultured, even though telomere shortening does not happen. This indicates again that oxidative stress is much larger phenomenon than telomere shortening. DNA damage in other than telomere region and mitochondrial dysfunction resulted by ROS may also contribute greatly to establish replicative senescence. ROS formed in mitochondria during ATP generation is one of inevitable byproducts prepared whenever we inhale oxygen. Cultivation of fibroblast in lower oxygen, instead of atmospheric oxygen resulted in delaying of replicative senescence. MEFs cultured in atmospheric oxygen contained more DNA oxidative damages than those cultured in lower oxygen, indicating that oxidative stress contributes to replicative senescence (Parrinello et al., 2003). Ascorbic acid captures directly or indirectly ROS and thereby relieves various oxidative stresses such as mitochondrial dysfunction, DNA damage, and telomere shortening. This relief of oxidative stress seems to result in the extension of replicative life span in fibroblast cells.
Why does ascorbic acid extend replicative life span of HEF cells only by 18% as shown in Fig. 2, even though ascorbic acid-treated HEF cells (PD 64) were similar to untreated young HEF cells (PD 30) rather than to untreated old HEF cells (PD 64) in ROS formation, mitochondrial damage and function, and AP site formation. One possible explanation is that water soluble ascorbic acid may not be enough to scavenge ROS in various cellular compartments. Other lipid soluble antioxidants like vitamin E may be necessary to have better efficiency in capturing ROS. However, this is not likely since co-treatment with ascorbic acid and vitamin E did not extend considerably replicative life span compared to treatment with ascorbic acid alone (data not shown). Another explanation is that intrinsic contributors that mediate partly replicative senescence but are not affected by oxidative stress appear to exist inside of cells. These intrinsic contributors will be deteriorated by cellular aging but not by oxidative stress. Therefore, although oxidative stress including changes in ROS level, mitochondrial damage and function, and AP site formation is relieved by antioxidants like ascorbic acid, intrinsic contributors that are not seriously related with oxidative stress but become less efficient during aging process will provide environment to cause replicative senescence. One possible intrinsic contributor will be cellular antioxidative enzymes and molecules like superoxide dismutase (SOD), catalase, peroxidase, glutathione, and so on. Although some of them are known to be activated by oxidative stress, their levels and activities are also reported to be decreased seriously during aging process. As we expected, age-related decrease in MnSOD and catalase activities in HEF cells have been reported (Li et al., 2006). The other possible intrinsic contributor will be DNA repairing system. Various DNA repairing activities have been reported to be diminished during aging process. Double strand break repair (DSBR) activity was decreased greatly during cellular senescence (Lombard et al., 2005; Vogel et al., 1999). Base excision repair (BER) activity was also known to be decreased seriously by aging process (Mostoslavsky et al., 2006). It is not clear yet that this reduction of DNA repairing activities during aging process is not related with oxidative stress. However, fibroblast cells obtained from long-lived mammals had longer replicative life span and generally higher DNA repair activities when compared to fibroblast cells obtained from short-lived mammals (data not shown, a manuscript is in preparation). There may be additional intrinsic contributors to help building up replicative senescence regardless of oxidative stress. On behalf of these intrinsic contributors, HEF cells still undergo replicative senescence, even though ascorbic acid extended somewhat their replicative life span by relieving oxidative stress. Replicative senescence is a complicated process that is mediated by various genetic and environmental contributors. Further detailed and systematical investigation will provide better understanding of replicative senescence by elucidating molecular mechanism.






 XML Download
XML Download