

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Riemerella anatipestifer (RA), once considered to be a member of the Pasteurella family, is now classified as a member of the Flavobacteriaceae family based on 16S ribosomal RNA (rRNA) gene analysis [29]. This Gram-negative bacterium causes septicemia and death in young ducks, geese, and other poultry worldwide [1523].
Continuous culture of bacterial pathogens in vitro can result in decreased virulence. In most cases, virulence is recovered in infection of sensitive hosts indicating that this phenomenon is not due to changes in the bacterial DNA. Thus, it can be hypothesized that altered virulence can be caused by transcriptional and translational changes in virulence factors.
To date, only five potential virulence factors have been reported for RA; outer membrane protein A (OmpA) [14], VapD [34], CAMP cohemolysin [4], TbdR1 [21] and siderophore-interacting protein (Sip) [32]. Among these, OmpA is an immunogenic protein involved in RA adherence [14]. The RA VapD gene shows homology with virulence-associated genes of other bacteria [34], and CAMP cohemolysin promotes the dissolution of red blood cells [4]. Sip significantly influences iron uptake and RA pathogenicity [32], while TbdR1 is involved in hemin and iron acquisition [21]. However, the molecular mechanisms involved in RA infection in vivo remain incompletely described.
Two-component signal transduction systems (TCSTs) are signal transduction mechanisms that exist widely in pathogenic bacteria. TCSTs respond to various environmental conditions and are composed of a sensor histidine kinase (HK) and a response regulator protein (RR) [35].
Recent research on RA has been limited to the in vitro investigation of the bacteria, while the processes involved in infection of ducks in vivo remain to be elucidated. It is conceivable that focusing on the growth and metabolism of RA in the host will be important not only in identifying new potential virulence factors but also in clarifying the pathogenic mechanisms of infection.
In this study, we used RNA sequencing (RNA-seq) as a sensitive and rapid method to determine transcriptional patterns and the sequence of the entire transcriptome [6] by comparing the total RNA obtained from bacteria grown both in vitro and in vivo. These results will provide insights into the mechanism underlying the pathogenesis of RA under in vivo conditions.
Materials and Methods
Bacterial strains, plasmids and culture conditions
The bacterial strains and plasmids used in this study are summarized in Table 1 [33]. The serotype 2 RA strain RAf153 was isolated from an infected domestic duck in Fujian Province, China. RA strains were cultured in tryptic soy broth (TSB; Difco Laboratories, USA) in a shaking incubator (180 rpm) at 37℃. E. coli strains X7213, X7213-pRE112 [10] and X7213-LSR-pRE112 were grown at 37℃ in Luria Bertani broth (LB) or plates supplemented with 50 µg/mL diaminopimelic acid (DAP). When necessary, the medium was supplemented with an appropriate concentration of antibiotics.
In vitro culture preparation
Strain RAf153 was inoculated into 5 mL TSB and cultured overnight. Bacteria (500 µL) were then transferred to 50 mL TSB and cultured to the logarithmic phase (optical density at 600 nm [OD600] 0.7–0.8). In vitro-cultured bacteria were harvested by centrifugation at 10,000 × g for 10 min at 4℃ and washed once with phosphate buffered saline (PBS). Three biological replicates were prepared.
Establishment of duck RA infection model
For virulence recovery, RAf153 were streaked onto tryptic soy agar (TSA) containing 7% sheep blood and cultured at 37℃ for 24 h. Single colonies of RAf153 were picked, inoculated into TSB, and cultured to the logarithmic phase (OD600 0.7–0.8). The bacteria were then harvested and washed with PBS three times before use in the in vivo challenge. The Cherry Valley ducks used in this study were purchased from a local duck farm. Cherry Valley ducks (aged 10 days) were injected intramuscularly with RAf153 at a dose of 1.5 × 109 colony-forming unit (CFU). After the appearance of symptoms such as lethargy, diarrhea, and tremor, infected ducks were humanely killed and the pathogen was isolated from heart and brain tissue. Considering the clinical signs of septicemia, in vivo conditions may provide a favorable environment for bacterial growth. The isolated bacteria were cultured in vitro and used to inject Cherry Valley ducks (aged 10 days) intravenously at a dose of 108 CFU. Infections were repeated in this way using doses of 107 CFU and 106 CFU. Eventually, we isolated the recovered virulence strain, vRAf153.
In this study, a model of acute RA infection was established in 2-week-old ducks challenged with a dose of 107 CFU of vRAf153 by plantar injection. Infected ducks showed symptoms such as lethargy, diarrhea, and tremor within 24 h. The levels of pathogens in the blood increased with exacerbation of the symptoms, with the highest levels reached immediately before death.
Identification and enrichment of in vivo cultured bacteria
RA-naïve ducks, according to indirect enzyme-linked immunosorbent assay results, were challenged on day 14 by plantar injection with a 0.2 mL inoculum (5 × 107 CFU/mL) of strain vRAf153 (OD600 0.7–0.8) [36]. The challenged ducks were closely monitored for 48 h post-infection. Cardiac blood (25–30 mL with added 3.8% trisodium citrate) was obtained before death. Giemsa staining and polymerase chain reaction (PCR) were used for detection and verification of RA.
Cardiac blood (20 µL) was streaked on TSA containing 7% sheep blood and cultured at 37℃ for 24 h. Single colonies were picked and inoculated into TSB for PCR identification. The primers were designed according to the 16S rRNA sequence named 16S rRNA-F and 16S rRNA-R (Table 2).
In order to evaluate the concentration of bacteria in blood, cardiac blood was 10-fold series diluted with PBS and cultured on TSB agar at 37℃ for 24 h. The number of bacteria were counted.
The bacteria in the collected blood were extracted by differential centrifugation according to the method described by Tan [30] with modifications. Briefly, three blood samples were centrifuged first at 200 × g for 10 min at 4℃; then, at 1,000 × g for 1 min and 1,500 × g for 30 sec to remove blood cells. The supernatant was centrifuged at 8,000 × g to collect bacteria. The purity of the RA extracted from blood was examined by Giemsa staining.
The animal experiments were approved by the Ethical Committee for Animal Experiments of Nanjing Agricultural University (Nanjing, China), and complied with the guidelines of the Animal Welfare Council of China.
RNA extraction
Total RNA was extracted by using the SV Total RNA Isolation System (Promega, USA) according to the manufacturer's protocols. RNA purity and integrity were assessed by measuring the A260/280 ratio and by 2% agarose gel electrophoresis. The RNA concentration was determined by using a NanoDrop2000 (Thermo Scientific, USA).
Six bacterial RNA samples comprising three biological replicates of both the in vivo and in vitro conditions were analyzed by RNA-seq at Guangzhou Genedenovo Biotechnology (China).
cDNA library construction and sequencing
The complementary DNA (cDNA) library construction and sequencing were performed by Guangzhou Genedenovo Biotechnology using a previously described protocol [19]. Briefly, the rRNA was removed, and the mRNA was extracted and fragmented by adding a fragmentation buffer. First-strand cDNA was then generated by randomly primed reverse transcription and the second-strand cDNA was synthesized using dNTPs, RNase H, and DNA polymerase I. The QIAquick PCR extraction kit (Qiagen, Germany) was used to purify the cDNA fragments. EB buffer was added for end reparation, poly(A) addition, and addition of sequencing adapters. After agarose gel electrophoresis and isolation of cDNA, the cDNA library was constructed by performing PCR amplification with the cDNA fragments as templates. Finally, the library was sequenced by using Illumina HiSeq 2000 (Illumina, USA). All raw transcriptome data were deposited in the GenBank Short Read Archive (accession No. SRP099396; National Center for Biotechnology Information, USA).
RNA sequencing data processing
In terms of the information provided by Guangzhou Genedenovo Biotechnology, raw reads generated by the sequencer are stored in a fastq file format. Impurity data including adaptor reads, low quality reads, and reads that contain unknown bases account for more than 10%. Clean reads are obtained by removing the impurity data from raw reads, and comprised over 95% of the total number of reads. Processing included comparing the clean reads to the reference sequence and analyzing the distribution. Based on the results of the comparison, the corresponding reads of each transcript are then calculated and standardized. The RPKM method was used to calculate gene expression levels.
Functional annotation and enrichment analysis
Based on gene expression, the relationships between samples and genes were performed by performing hierarchical clustering, and heat maps were used to present the clustering results. The genes with similar expression patterns usually have functional correlation. The differentially expressed genes (DEGs) were used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses via the GO and KEGG databases, respectively. GO is an internationally standardized gene functional classification system that provides a dynamically updated controlled vocabulary to comprehensively describe the properties of genes and gene products in organisms. GO has three ontologies, describing molecular function, cellular components, and biological processes. In vivo, different genes coordinate and perform their biological functions, and analysis based on pathways will help to elucidate the biological function of genes. KEGG is the main public pathway database. Enrichment analysis of pathways is based on the KEGG pathway database, and the hypergeometric test is used to identify the pathway in which DEGs are significantly enriched.
Quantitative real-time PCR validation
RNA samples similar to those sent for RNA-seq analysis were reverse-transcribed into cDNA in a total reaction volume of 20 µL by using the Vazyme HiScript Q RT SuperMix (Vazyme, China) for qPCR (+gDNA wiper). The reactions were as follows: 25℃ for 10 min, 50℃ for 30 min, and 85℃ for 5 min. Completed reaction products were collected and pooled before 20 µL aliquots were stored at −70℃. The cDNAs were diluted 1:50 before use in quantitative real-time PCR (qRT-PCR) analysis to confirm the accuracy of the RNA-seq data. Six DEGs identified by RNA-seq were selected as templates for the qRT-PCR assays. Using the cDNA from RAf153 cultured in vivo and in vitro as templates, three replicates were prepared for each reaction. Primers for qRT-PCR were designed by Primer Express software (ver. 3.0; Applied Biosystems, USA). The gene gmk (GI:874285954) was used as an internal control, and relative quantification was calculated based on the 2−ΔΔCt method [20].
Construction of the mutant strain ΔArsR-SthK
Briefly, genomic DNA was isolated from vRAf153 and vector pSET4s (containing the spectinomycin resistance cassette, spcR cassette). Primers for the amplification of upstream (ST-L1, ST-L2) and downstream (ST-R1, ST-R2) fragments of the target gene and the spcR cassette (Spc-1, Spc-2) are listed in Table 2. After amplification and purification, these three fragments were fused by overlap-extension PCR with the spcR cassette located between the upstream and downstream fragments of the target gene.
E. coli X7213-pRE112 was cultured in LB medium containing 50 µg/mL DAP and 25 µg/mL chloramphenicol (Cm) and shaken overnight at 37℃. The pRE112 and fusion segment were digested with Kpn I and Sac I and ligated overnight at 16℃ to generate the recombinant plasmid LSR-pRE112. Competent cell E. coli X7213 cells were transformed with LSR-pRE112 and cultured on LB plates supplemented with 50 µg/mL DAP at 37℃ for more than 24 h. Single colonies were picked for sequencing and positive clones were identified as the donor strain, designated E. coli X7213-LSR-pRE112.
The recipient strain vRAf153 was cultured in TSB and the donor strain was cultured in LB medium (supplemented with 50 µg/mL DAP, 50 µg/mL Spc, and 25 µg/mL Cm) until the culture reached an OD600 value of 0.6 to 0.8. Aliquots (5 mL) of the donor and receptor strains were centrifuged at 5,000 × g for 3 min and washed three times with 1 mL 10 mmol/L MgSO4. After the third wash, the supernatant was removed and the pellets were combined. The mixed cultures were then incubated on a TSB plate containing 50 µg/mL DAP at 37℃ for 10 h. The bacteria on the plate were washed with TSB and streaked onto a TSB plate containing Spc (50 µg/mL) to isolate the putative vRAf153 mutants from the mixed-strains culture. Single colonies were selected from the plate and the candidate mutants were screened and identified.
Validation of gene-deleted strain ΔArsR-SthK
PCR amplification was performed with ΔArsR-SthK as a template and ST-L1 and ST-R2 as primers. The PCR products were extracted from 1% agarose gel and were sequenced by Guangzhou Genedenovo Biotechnology. The amplified fragments were blasted and aligned to the genome of RAf153 to make sure that the SpcR cassette and genes ArsR and Sthk were replaced via allelic exchange.
Internal primers (inST-1 and inST-2 in Table2) of the deleted genes were designed to verify that the genes ArsR and SthK had been deleted. Primers inSpc-1 and inSpc-2 (Table 2) were used to confirm that the resistance gene has been inserted. Moreover, ΔArsR-SthK was further validated by primers 16S rRNA-F and 16S rRNA-R (Table 2).
In addition, according to the whole genome sequence published on GeneBank, the upstream and downstream genes of ArsR and Sthk were CG09_1826 and CG09_1830. Internal detection primers (up-1, up-2 and down-1, down-2 listed in Table 2) were designed. The cDNA of ΔArsR-SthK and RAf153 were amplified to verify whether the deleted fragments affect the upstream and downstream genes.
Adhesion and invasion assays
Adhesion and invasion assays were performed to determine whether the deletion of ArsR and SthK could affect the adhesion and invasion abilities of RA. Briefly, Vero cells were seeded into 24-well plates and incubated to 95% confluence. Strains vRAf153 and ΔArsR-SthK were cultured to mid-log phase (OD600 = 0.7–0.8) and Vero cells were infected at a multiplicity of infection of 50, centrifuged (800 × g for 10 min), and incubated at 37℃ with 5 % CO2 for 2 h. Cells were washed three times by PBS to remove non-adherent bacteria. For the adherence assay, cells were lysed by using sterile water. Cell lysates were 10-fold series diluted and spread on TSB agar plate to count the number of bacteria. For the invasion assay, extracellular bacteria were killed by adding DMEM containing 5 µg/mL penicillin and 100 µg/mL gentamicin and incubated for 1 h. Then, after three washes, cells were lysed and spread on TSB agar to determine the quantity of bacteria. Both assays mentioned above were performed in triplicate and repeated three times.
Determination of LD50 of vRAf153 and ΔArsR-SthK
Five-day-old Cherry Valley ducklings were obtained from a local duck farm. After 2 days of acclimatization, the ducklings were divided into 11 groups (n = 6 per group). The vRAf153 strain and the mutant strain ΔArsR-SthK were grown in TSB at 37℃ in a shaker and harvested when the culture reached an OD600 value of 0.7 to 0.8. The cultures were then centrifuged at 6,000 × g for 2 min and resuspended in PBS at the appropriate densities. Ducks were injected intramuscularly with vRAf153 or the deletion mutant strain at the doses listed in Table 3. The 50% lethal dose (LD50) was calculated by using the Reed-Muench formula [24].
Statistical analysis
For RNA-seq results, the significance of differential gene expression was analyzed by using a threshold for the false discovery rate of ≤ 0.005 and an absolute value of the log2 ratio ≥ 1. For the qRT-PCR data analysis, a p value less than 0.05 was considered statistically significant.
Results
Bacterial extraction from in vivo cultures
Approximately 20 h after challenge with vRAf153, blood was obtained by exsanguination from the heart immediately before death. Bacteria presence in the blood was determined by Giemsa staining and visualization via light microscopy (panel A in Fig. 1; left). RA pathogens in vivo were collected by differential centrifugation and high purity was confirmed by Giemsa staining (panel A in Fig. 1; middle). In addition, RA in blood was identified by performing PCR (panel B in Fig. 1), which confirmed the bacteria isolated from the blood were RA.
The blood from the heart was diluted prior to counting the bacteria as the number of colonies per milliliter of blood can reach 109 CFU. In general, there were 1010 CFU of bacteria extracted from the blood, and one-third of the bacteria sample was used for each RNA sample preparation.
GO and KEGG analyses of DEGs
By comparison with bacterial genes expressed under in vitro conditions, we identified 682 (682/2,101, 32.46%) upregulated genes (Supplementary Table 1) and 121 (121/2,101, 5.76%) downregulated genes (Supplementary Table 2) under in vivo conditions (Fig. 2).
To gain additional insight into the expression of genes by RAf153 cultured in vivo, all DEGs were classified into three GO categories: biological process, cellular component, and molecular function (Fig. 3). As summarized in Supplementary Table 3, cellular and metabolic processes were the dominant subcategories in the biological process category, cell and cell part were the most abundant in the cellular component category, and catalytic activity and binding were highly represented in the molecular function category.
In KEGG pathway annotation, 313 DEGs were represented among the pathway annotations, with metabolic pathways, biosynthesis of secondary metabolites, and ribosomes (62, 29, and 19, respectively) accounting for the majority of annotations (Supplementary Table 4).
In addition, a heatmap of expression was constructed for those genes that showed significantly different expression between the in vivo and in vitro culture conditions (Fig. 4). In Fig. 4, each column represents an experimental sample, each row represents a gene, and the expression levels of the gene in different samples are shown in different colors; red indicates an upregulated expression level and green indicates a downregulated expression level.
qRT-PCR analysis
Six genes were selected as templates for validation of the transcriptomic data by qRT-PCR analysis, and the reactions were performed in triplicate. The qRT-PCR results were largely consistent with those obtained from the RNA-seq analysis (Fig. 5), which confirmed the validity of the Illumina sequencing data.
Significant KEGG pathway enrichment
Pathway enrichment analysis reflects the metabolic changes of pathogenic bacteria during culture. Some significantly upregulated genes were identified in three pathways (Table 4) involved in virulence, protein secretion, and signal transduction.
Characterization of ΔArsR-SthK
Primers ST-L1 and ST-R2 were used to amplify ΔArsR-SthK, and the target fragment was sequenced for BLAST analysis. Compared to the genes of RAf153, the genes ArsR and SthK on strain ΔArsR-SthK were deleted (panel A in Fig. 6; upper). Moreover, panel A in Fig. 6 (lower) indicated that the spcR cassette was integrated into the genome of ΔArsR-SthK. Thus, the spcR cassette and the genes ArsR and SthK were allelically replaced, and the deleted strain ΔArsR-SthK was constructed. The mutant strain was further validated by primers inST-1 and inST-2, inSpc-1 and inSpc-2, 16S rRNA-F and 16S rRNA-R (panel B in Fig. 6).
In addition, the upstream and downstream fragments of deleted genes were amplified through qRT-PCR, and the results in panel C in Fig. 6 show that upstream and downstream genes of ArsR and Sthk underwent transcription normally and that a polar effect did not occur.
Adhesion and invasion ability of vRAf153 and ΔArsR-SthK on Vero cells
The results suggested that deletion of genes ArsR and SthK decreased the capacity for adhesion and invasion significantly (Fig. 7), which suggested that genes ArsR and SthK may be involved in RA's infection of host cells.
Virulence of vRAf153 and ΔArsR-SthK in ducklings
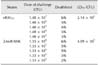
To evaluate the virulence of the mutant strain, the LD50 of the vRAf153 and mutant ΔArsR-SthK strains were determined in Cherry Valley ducklings. Observation of mortality of ducklings for 7 days post-challenge revealed that the LD50 value for the mutant strain was more than 19 times higher than that for vRAf153 (4.09 × 105 CFU vs. 2.14 × 104 CFU, respectively; Table 3). These data indicated that inactivation of ArsR and SthK reduced bacterial virulence.
Discussion
In this study, we identified a panel of DEGs for RA cultured in vivo and in vitro by using RNA-seq. With regard to RAf153, 2101 of 2157 genes were identified, with a coverage rate of 97.68%. BLAST searching showed two of the upregulated genes, ArsR and SthK, located on a single operon and revealed that these genes form a two-component system. The function of these genes in bacterial virulence was then investigated by generating a knockout mutant strain. Animal experiments showed decreased virulence of the mutant compared to that of the wild-type strain, thus implicating ArsR and SthK as potential RA virulence genes.
By comparison with bacterial genes expressed in vitro, we identified 682 genes that were upregulated under in vivo culture conditions. Some of these genes were enriched in one KEGG pathway, and some were reported to be related to virulence or to enabling the pathogen to survive and multiply in vivo.
The upregulated DEGs enriched on pathway KO00270 were related to methionine biosynthesis, which is critical for replication and infection and has been shown to be required for the full virulence of Brucella melitensis [17] and Haemophilus parasuis [12]. The methionine transport system is also critical for group B Streptococcus survival in vivo [28]. The DEGs enriched on pathway KO02010 related to ATP-binding cassette (ABC) transporters were upregulated under in vivo culture conditions. In bacteria, ABC transporters play a significant role in the uptake of essential nutrients, such as iron, heme, and vitamin B12, and are also important in the clearance of toxins and drugs [1322]. Upregulated DEGs were also identified in pathway KO02020 related to two-component systems.
The urease gene cluster of RAf153, containing the structural genes encoding urease alpha, beta, and gamma subunits as well as accessory genes (ured, ureg, and uref), are homologous to other bacterial urease genes. The urease beta and gamma subunits were significantly upregulated in bacteria cultured in vivo. Urease functions as a virulence factor in several pathogenic bacteria, such as Helicobacter pylori, and Klebsiella aerogenes [26]. Although this function for urease has not been previously reported, the upregulation of urease gene cluster genes identified in this study implicates urease as a virulence factor in this strain. However, this speculation requires further investigation. Upregulation of transcriptional regulators is important in host acclimatization to various environmental and physiological stresses. In bacteria, the AraC family of transcriptional regulators are reported to be involved in carbon metabolism, stress responses, and virulence regulation [9]. Furthermore, mutation of the AraC family transcriptional regulator Rv1931c suggested its roles in regulating the expression level of genes involved in the virulence of Mycobacterium tuberculosis [8]. The expression of four genes, orf02176, orf02155, orf01543, and orf00132, annotated as CRP/FNR family transcriptional regulators, tended to increase in vivo. In Enterococcus faecalis, the transcriptional regulator Ers, which is a member of the CRP/FNR family, was reported play a role in oxidative stress responses as well as in bacterial virulence [25]. Furthermore, the CRP/FNR family protein Bcam1349 binds the second messenger cyclic diguanosine monophosphate (c-di-GMP) to regulate biofilm formation and virulence in the pathogenic strain Burkholderia cenocepacia [7]. Thermusther mophilus SdrP, which is also a member of the CRP/FNR family proteins, alters the expression of genes in response to a variety of signals, such as cAMP concentration and anaerobic conditions [1]. Previous studies have shown that CRISPR-Cas systems function in the defense of bacteria against foreign nucleic acids. The Francisella novicida Cas9 protein has a critical role in evading the host immune response and promoting bacterial virulence, and mutation of CRISPR-Cas proteins restores the ability of the host to mount an effective immune response, thus representing a strategy that may also applied to other pathogens [27]. According to our RNA-seq analysis, CRISPR clusters encoding Cas proteins 1, 2, and 9 were all upregulated under in vivo culture conditions, which indicates the involvement of the CRISPR-Cas systems as virulence factors in RA infection. Upregulated DEGs were identified in relation to biosynthesis of peptidoglycans, which are essential for maintaining shape in strains such as Leptospira interrogans in which peptidoglycans provide sufficient strength and flexibility to withstand osmotic pressure [2]. An intact cell wall is required for full bacterial virulence.
By comparison with bacterial genes expressed in vitro, we identified 121 genes that were significantly downregulated under in vivo culture conditions. These included genes involved in protein synthesis, which is consistent with the DEGs identified in Actinobacillus pleuropneumoniae cultured under in vivo conditions [5]. Nearly 40 downregulated genes were enriched on pathway KO03010 related to ribosomes, which are required for protein synthesis. Among these DEGs, 37 were 30S and 50S ribosomal proteins showing downregulation ranging from −1.02-fold to −3.15-fold. The specific regulation mechanism of this process remains to be fully elucidated. The expression of molecules involved in the protein folding process were also downregulated. Trigger factor chaperones are responsible for regulating the folding of small proteins, while complete folding of some larger proteins requires DnaK and DnaJ, and encapsulated proteins are further folded by the GroEL and GroES system [331]. Furthermore, groel is highly conserved among different RA strains, and Chaperonin GroEL has been identified as an immunogenic protein in different serotypes of RA [11]. Thus, genes encoding trigger factors, DnaK, GroEL, GroES, and GrpE, which are important in the DnaK chaperone system, were all downregulated in RA cultured under in vivo conditions.
TCSTs are widely used signal transduction mechanisms that respond to various environmental conditions in both Gram-positive and Gram-negative pathogenic bacteria. These systems are composed of a sensor HK protein located on the cell membrane and a RR protein located in the cytoplasm [1635]. The HK monitors environmental changes, while the RR conveys the signals from HK to regulate gene expression in response to environmental changes. In this study, eight upregulated DEGs were annotated to pathway KO02020 related to TCSTs. These included genes encoding structural components, with ArsR coding signal transduction RR and SthK coding the protein HK. TCSTs play major roles in regulation of virulence and pathogenesis [18]. Evaluation of a mutant containing a deletion of the ArsR and SthK genes, which are located in the same operon, showed decreased virulence compared with that of the wild-type strain. This result indicates that the ArsR and SthK genes represent the signal transduction HK and signal transduction RR genes in RA, indicating their potential as virulence factors.
In this study, we established a duck model of acute RA infection and conducted comparative transcriptome analysis to provide new insights into the mechanism of RA pathogenesis in vivo. The ArsR and SthK knockout mutant revealed the essential role of these genes as an effective signal transduction system in bacterial virulence. Further analysis of DEGs is required to fully elucidate some of the adaptations that occur in RA during in vivo infection.




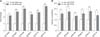





 XML Download
XML Download