

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The cellular plasma membrane restricts the internalization of macromolecules from the outside environment. As such, several methods have been developed to facilitate the delivery of macromolecules into cells such as the use of liposomes, electroporation, and viral transfection. Cell-penetrating peptides (CPPs), which are generally composed of 4~30 basic amino acid rich sequences, are promising tools for delivering macromolecules into the cells (123).
In 1965, it was demonstrated that basic amino acid-rich histones and basic poly-amino acids can stimulate the internalization of albumin into tumor cells (4). Later studies showed that conjugation of poly-L-lysine to albumin enhanced cellular uptake (5). In 1988, cell membrane penetration of trans-acting activator of transcription (TAT) protein from human immunodeficiency virus-1 (HIV-1) was observed (6). It was shown that the arginine and lysine rich domain of TAT was critical for membrane penetration (7), and covalent binding of this domain with other proteins induced cellular uptake of proteins (8). These studies led to the identification of a number of CPPs including VP22, a CPP derived from viral protein (910); penetratin derived from the antennapedia, a drosophila homeoprotein (1112); and transportan, derived from a neuropeptide (13). In addition to these natural sequences, synthetic CPPs such as poly-arginine (14), model amphipathic peptide (MAP) (15), and TP2 (16) were also generated. These CPPs are internalized across the plasma membrane alone or together with macromolecules such as proteins and nucleic acids.
The mechanism of internalization is still not fully understood. However, it is known that small CPPs can be internalized by both endocytosis and direct translocation across the membrane whereas large CPP-cargo molecules are internalized only via the endocytic pathway (21718). After internalization, the CPP-cargo molecules enclosed in endosomes fuse with the lysosome. However, due to as yet unknown natural properties of some CPPs or modifications such as conjugation with chemicals escaping from the endosome or amino acid sequences, CPPs can escape from the endosome and diffuse into the cytoplasm before lysosome fusion (119). Because of the basic amino acid-rich characteristic of CPPs, similar to the nuclear localization sequence (NLS), some CPPs can translocate into the nucleus and as such, transcription factors and plasmid DNAs are often used as cargo for CPPs (2021).
Various therapeutic approaches have been applied using CPPs (21722). Delivery of biologically active proteins is the most common approach. Chemical conjugation or CPP sequence addition at the N- or C-terminal of the protein induces cell internalization. Addition of an 11 amino acid TAT sequence to the N-terminal of 116-kDa β-galactosidase (β-gal) led to in vitro and in vivo cellular internalization of the recombinant TAT-β-gal (8). Owing to their positively charged amino acid-rich characteristics, CPPs can bind to negatively-charged nucleic acids (23). Several studies have explored delivery of plasmid DNA (pDNA) using CPPs and a simple mixture of pDNA with TAT facilitated the transfection of pDNA into cells (2425). CPPs can also be used to transfect siRNA into cells (2627). However, the transfection efficiency of CPP/nucleic acid complexes is low, and various methods have been developed to overcome this limitation, such as the addition of chloroquine, which induces endosomal escape, (28) and using CPP-conjugated RNA binding domain (RBD) for RNA delivery (2930). Also small molecules such as taxol and methotrexate conjugated with CPPs showed improved drug efficiency. Because of decreased transport and increased efflux. It was reported that various tumor cells showed resistant to chemotherapy while CPP-conjugated small molecules increased drug delivery into cells and showed better therapeutic efficiency to tumor cells (3132).
From the cargo delivery properties, there are CPP-based drugs undergoing clinical trials. TAT-linked c-Jun N-terminal kinase (JNK)-inhibiting peptide, called XG-102, is undergoing clinical phase III that targeted different diseases such as inflammation and hearing loss. KAI-9803 and KAI-1678 are TAT-CPP conjugated protein kinase C inhibitor peptide for myocardial infarction and pain. Clinical phase II for these molecules were completed. TAT-CPP using botulinum toxin type A, named RT001, is currently in clinical phase III for treatment of wrinkles (17). Besides these molecules, various molecules using CPPs have been underwent or are currently in clinical phase but there are no FDA approved molecules yet.
One of the most promising therapeutic approaches using CPP is its application in dendritic cell (DC) vaccine based treatment of cancer and infectious diseases. The predominant method of DC immunization involves the use of antibodies or molecules that target surface receptors such as C-type lectin receptors, FC receptors, and integrin. After interaction with these receptors, conjugated antigens are internalized by endocytosis. The endosome with the antigens is fused with the lysosome and antigens are degraded by protease, and then loaded onto MHC class II. Moreover, some DCs can cross-present antigens on MHC class I to CD8+ T cells; however, the mechanism is not clearly understood. Endocytosed antigens can also escape into the cytoplasm, undergo proteasome degradation, and ultimately transported to the endoplasmic reticulum (ER) (3334). Accordingly, CPP-antigens can be internalized into DCs and loaded onto MHC molecules. Because CPP-antigens can be internalized into DCs without receptor targeting molecules, this method is independent of receptor expression. Also, the natural propensity for endosome escape demonstrated by some CPPs enable antigen presentation by MHC class I molecules, leading to more efficient CTL responses than the naked antigen pulsing method. In this review, we highlight various CPP applications for immune modulation including DC-based vaccination, and discuss the advantages of using CPPs in dendritic cell biology.
CPP APPLICATIONS IN IMMUNE MODULATION
Several approaches to immune modulation using CPPs have been attempted. One of the initial strategies was delivery of dominant-negative signaling molecules that can competitively inhibit the function of endogenous proteins in immune cells. Ras is an important signaling protein for Th2 differentiation after TCR stimulation (35). Intranasal delivery of dominant-negative Ras (dnRas) using TAT-CPP blocked OVA induced eosinophilia and lymphocyte accumulation in the bronchoalveolar lavage (BAL) fluid. Lung histology demonstrated a decrease in cellular infiltration and mucus-containing epithelial cells in TAT-dnRas pretreated mice (36). Phosphoinositide 3-kinase (PI3K) signaling is an important pathway down-stream of the costimulatory molecules in T cells and B cells, which contributes to asthma by phosphorylation of PtdlnsP2, immune cell recruitment, activation, and apoptosis. PI3K is composed of a catalytic subunit and adaptor protein. p85α is a PI3K adaptor protein and p85α knockout mice die within a few weeks of birth due to immunodeficiency (37). After intraperitoneal injection of dominant-negative p85α, which lacks the binding site for the catalytic subunit conjugated to TAT-CPP, lymphocyte and eosinophil numbers were reduced. In addition, the level of IL-4 and IL-5 decreased in the BAL fluid in OVA immunized mice (38). ZAP-70 is a proximal TcR signaling molecule that induces phosphorylation of downstream molecules such as LAT and SLP-76 (39). Mutation of tyrosine 318 (Y318F mutant), which is the amino acid for phosphorylation, impairs T cell activation signaling. Treatment of dominant negative ZAP-70-Y318F conjugated with human derived CPP, named Hph-1, reduced phosphorylation of TcR signaling and inhibited secretion of IL-2 after TcR stimulation (40). STAT-6, a cytokine receptor that regulates IL-4/IL-13 gene expression, is also critical to allergic airway diseases and T cells from STAT-6 knockout mice cannot be differentiated into Th2 cells (41). Intranasal administration of a TAT-CPP conjugated dominant-negative STAT-6 peptide induced the asthma model in mice through OVA stimulation. Moreover, there was a significant decrease in the number of immune cells from BAL fluid and mucus production in the lung (42). Retinoic acid-related orphan receptor gamma t (RORγt) is a lineage specific transcription factor for Th17 differentiation (43). Dominant negative truncated form of RORγt without its DNA binding conjugated with Hph-1-CPP showed reduced IL-17 production and Th17 differentiation results amelioration of experimental autoimmune encephalomyelitis (EAE) (44).
In addition to delivering dominant negative molecules into cells, intracellular delivery of negative regulators of key signaling pathways of the immune system was another strategy utilized for CPP mediated immune modulation. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is expressed in activated T cells and regulatory T cells, and functions to down-regulate the immune system (45). Treatment with the cytoplasmic domain of CTLA-4 (ctCTLA-4) recombinant protein conjugated to Hph-1-CPP inhibited IL-2 secretion from activated T cells. Intranasal administration of Hph-1-ctCTLA-4 reduced Th2 cytokines as well as the number of immune cells from BAL fluid. Inhibition of inflammatory cell infiltration and mucus containing epithelial cells was also observed in an OVA induced mouse asthma model (46). Intravenous injection of Hph-1-ctCTLA-4 also inhibited inflammation and bone destruction in a collagen induced autoimmune arthritis model through inhibition of T cell responses (47). Another application involved intravenous injection of ctCTLA-4 conjugated with dNP2-CPP, which can penetrate the blood-brain barrier and deliver the protein into the brain and spinal cord in an experimental autoimmune encephalomyelitis (EAE) model. This resulted in reduced IFN-γ or IL-17A producing CD4+ T cells in the spinal cord, and ameliorated EAE (48). In other study, suppressor of cytokine signaling 3 (SOCS3) a repressor of IL-6 induced phosphorylation of STAT3 was linked with MTM-CPP and it showed inhibition of inhibited inflammatory liver injury induced by enterotoxin B (SEB), lipopolysaccharide (LPS), and lectin concanavalin A (ConA) (49).
Foxp3, a master transcription factor for regulatory (Treg) T cells, was generated as a recombinant protein conjugated to tandem repeated form of Hph-1-CPP, named HHph-1. It could be translocated into the nucleus and convert CD4+ CD25- T cells to Treg like CD25highCTLA-4high cells. In addition, systemic delivery of HHph-1-Foxp3 suppressed allergic airway inflammation and autoimmune responses in a colitis model and in scurfy mice (50).
Chimeric peptide based immune modulation by targeting transcription factors has also been attempted. NF-κB is an important transcription factor that regulates multiple genes related to immune responses (51). In pharmaceutical studies, targeting NF-κB is a popular strategy for regulating immune responses. NF-κB essential modulator (NEMO) is a subunit of the IκB kinase complex (IKK) that activates NF-κB through phosphorylation (52). The NEMO binding domain (NBD) is an 11-amino acid peptide that can block the interaction of NEMO with IKK and inhibit NF-κB activation. NBD conjugated to TAT-CPP inhibited LPS-induced IKK activity, accelerated constitutive apoptosis of human polymorphonuclear neutrophils (PMNs), and inhibited LPS-delayed apoptosis (53). Nuclear factor of activated T cells (NFAT) is a key transcription factor for T cell activation and IL-2 production (54). The VIVIT peptide was identified as an inhibitor for NFAT signaling through the interfering interaction of calcineurin with NFAT (55). Intranasal delivery of a Sim-2-VIVIT peptide inhibited airway inflammation in an OVA-induced experimental asthma model (56). Mitogen-activated protein kinase activated protein kinase 2 (MK2) is a serine/threonine kinase that regulates cytokine expression at the transcriptional and post-translational levels by stabilizing the mRNA of inflammatory cytokines (57). The MK2 inhibitory peptide, KKKALNRQLGVAA, conjugated to various CPPs blocked the production of inflammatory cytokines such as IL-6 and TNFα in LPS stimulated macrophages (58).
Nucleotides have also been delivered into cells using CPPs for immune regulation. When siRNA targeting mitogen-activated protein kinase (MAPK) was conjugated to TAT-CPP or penetratin-CPP and delivered into the cells, MAPK mRNA levels were reduced by siRNA-CPP in vitro; however, intratracheal administration of siRNA-CPP did not affect MAPK mRNA levels in vivo (59). Decoy oligonucleotides that consist of a double-stranded short oligonucleotide corresponding to a gene promoter can block the binding of transcription factors to promoter regions. A NF-κB decoy was hybridized to the peptide nucleic acid (PNA) and PNA was linked to transportan-CPP by disulfide bond. Following intracellular delivery, this complex blocked NF-κB binding activity and decreased IL-1β induced IL-6 mRNA (60).
Methotrexate (MTX) is a small molecule drug for treatment of rheumatoid arthritis. However, the long-term systemic administration of MTX may induce serious adverse effects. In this reason, previously MTX conjugated with cell-penetrating peptide, Hph-1, was used for transcutaneous topical administration for rheumatoid arthritis. This cell-penetrating MTX showed successful therapeutic effects in collagen induced arthritis model by inhibiting pro-inflammatory cytokine expressions and inflammation (61). Cyclosporine A (CysA) was also conjugated with poly-arginine (R7) to enhance its delivery efficiency in vitro and in vivo, especially topical skin delivery. It showed successful inhibition of T cell activation and ear inflammation (62).
As we summarized, various therapeutic cargo molecules including proteins, peptides and nucleotides have been applied in immune disease models targeting key functional molecules in immune cell activation and differentiation. There have also been various clinical trials under investigation for the commercialization of CPP conjugated bio-drugs in immune diseases (17). Biologic drug targeting of intracellular events using CPP could open a new area of investigation into drug development.
DC VACCINATION WITH CELL-PENETRATING PEPTIDES
Conventional DC vaccination method
Dendritic cells are the major mediators between innate and adaptive immune responses. They can initiate antigen specific adaptive immune responses in their role as antigen presenting cells (63). Because of this critical function, they are considered potent immunotherapeutic agents, especially for treating various cancers (64). In 1995, the first attempt at ex vivo DC-based cancer vaccination was reported for the treatment of melanoma (65), and since then, numerous clinical trials using DC vaccination have been completed or are currently underway.
ex vivo DC manipulation is the most common methodology used in clinical trials. This requires the isolation of DCs or their precursors from patients or donors (66). The sources of DCs are autologous or allogeneic cells. Autologous cells have the advantage of no risk of graft-versus-host disease but allogeneic cells could induce efficient immune boosting (6768) because the isolated cells may have immune suppressive characteristics (6970). After obtaining the cells, manipulation is performed, including appropriate antigen loading (71), maturation, and activation (7273). Successfully manipulated DCs are then reinjected into patients by various routes including intradermal/subcutaneous (74), intranodal (75), intratumoral (7677), or intravenous (78) injection. For example, to treat melanoma, mature DCs (79), CD34+ cells (80), or monocyte-derived DCs (81) pulsed with peptide antigen showed significant immune responses. In addition to using peptide antigen, killed allogeneic tumor cells were also used as antigens and they demonstrated successful immunogenicity, clinical responses and prolonged survival (82). DC vaccination approaches have been used to treat other types of cancer including prostate cancer (83), colon cancer (84), or glioma (85), and multiple myeloma (86), and have demonstrated successful immunogenicity and T cell responses.
Another methodology for DC vaccination is the in vivo DC targeting approach. Because ex vivo DC manipulation and reinjection in patients is generally regarded as an expensive and laborious process (66), the injection of antigen coupled to DC cell surface targeting monoclonal antibodies (mAb) has been applied. The use of DEC205 (87), DC surface lectins DCIR (88), dectin 1 (89), CLEC9A (90), and Langerin (91) targeting antibodies coupled to antigens was evaluated. Animal studies demonstrated that antigen delivery using these molecule specific antibodies induced antitumor immunity involving T cell responses and antigen specific antibody responses. This approach was also used for anti-viral vaccination using an anti-DEC205 antibody coupled to the HIV gag protein (92). This approach was also successful, demonstrating induction of robust T cell immunity in nonhuman primates. However, this method can only be used with known antigen because the cells need to be prepared experimentally prior to injection. This is in contrast to ex vivo DC manipulation that utilizes tumor cell lysates or apoptotic cell bodies as antigen. Also, to induce efficient DC activation and robust adaptive immunity, adjuvant is needed (93). Furthermore, the impact of the interaction between cell surface molecules on DCs and targeting mAbs is carefully characterized to induce only accurate and expected immune responses (94).
DC vaccination using cell-penetrating peptides
Although ex vivo DC manipulation has demonstrated clinical success, its efficiency has not been regarded as effective. It has been reported that enhancing antigen delivery efficiency and long lasting antigen presentation could improve this weakness by using cell-penetrating peptide conjugated antigens. For example, CPP1 (AAVLLPVLLVLLAAP) conjugated peptide antigen pulsed DCs induced successful anti-tumor responses in vivo with much higher efficiency than only antigen pulsed DCs (95). This suggests that CPP mediated antigen delivery in the cytoplasm facilitates antigen presentation by newly synthesized MHC class I molecules, which lead to long-term immunological responses.
Accordingly, one of best-characterized CPPs, TAT, has been used as part of various DC vaccination strategies. TAT fused Leishmania homolog of receptors for activated C kinase (LACK) was used to induce immune responses against leishmaniasis through DC vaccination (96). TAT-LACK pulsed DCs induced higher proliferation of CD8+ T cells and IFN-gamma releasing Th1 or Tc1 cells than LACK only pulsed DCs, suggesting that TAT enhanced vaccination efficiency. In another study, the OVA protein fused with TAT (TAT-OVA) induced antigen specific cytotoxic lymphocytes (CTL) while OVA lacking TAT could not enter the MHC class I presentation pathway (97). In a breast tumor model, mature TAT-Her/neu pulsed DCs induced Her2/neu specific CD8+ T cell responses and CD4+ T cell responses (98).
Another CPP, called penetratin (Antp), a 16-mer peptide, has also been used for DC vaccination, and its efficacy has been confirmed through in vitro and in vivo experiments (99). DCs pulsed with penetratin linked to the CD4 or CD8 specific OVA epitope for 24 h and co-cultured with OT-I or OT-II cells successfully induced T cell proliferation. In addition, in vivo injection of the penetratin-antigen complex induced an immune response against OVA expressing tumor cells, and its ability to reduce tumor size was enhanced through adjuvant co-injection. Poly-arginine (R9) has also been efficiently applied in DC vaccination strategies, in terms of inducing antigen specific T cell proliferation and reducing tumor size (100). Recently, a new CPP named, z12 was used for direct antigen delivery in vivo to dendritic cells, and showed robust CD4+ and CD8+ T cell mediated antitumor immune responses (101).
Collectively, these studies suggest that CPP can be used for DC vaccination against infectious diseases or various cancers. Although CPPs can only be coupled to known antigens for preparing fusion proteins or peptides, CPP-antigen delivery efficiency and location in the cytoplasm can significantly enhance the efficiency of cytotoxic T lymphocyte (CTL) activation, presumably via cross-presentation. This DC vaccination method has been shown to be more effective than naked antigen pulsing methods.
Mechanisms of CPP-antigen delivery and processing
CPP can interact with cell surface molecules such as glycosaminoglycans (GAGs), which are negatively charged-cell surface glycoproteins (102103). Most well characterized CPPs are positively charged amino acid based sequences. Under physiological conditions, their electrostatic interaction with GAGs can easily be formed, and this CPP-cell surface molecule interaction induces internalization of cargo molecules into the cytoplasm. The internalization mechanisms are defined as macropinocytosis or clathrin-, caveolin-mediated endocytosis, which lead to the formation of endosomes (17). In DC vaccination using CPPs, these mechanisms significantly enhance antigen delivery efficiency compared to free antigen delivery systems, and mimic the antigen presentation pathway of naturally phagocytosed antigens.
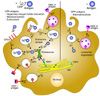
Because internalized CPP conjugated antigens in DCs are in the endosome, they can be encountered by two pathways: MHC class II presentation or MHC class I presentation by cross-presentation (104). When DCs uptake an antigen, the phagocytosed endosome fuses with the lysosome and the antigen is then degraded into peptides. In this late endosome, antigens can be loaded onto MHC class II and presented on the surface of DCs. CD4+ T cells can recognize this MHC class II-antigen complex for inducing adaptive immune responses. Also, in terms of DC vaccination, MHC class I cross-presentation is an important process for recruiting CD8+ T cell responses against specific antigens. Antigens that escape from endosomes are degraded through proteasomal degradation and can be loaded on MHC class I in cytosolic vesicles through the transporter associated with antigen processing 2 (TAP2) molecule. Degraded antigen peptides can also be transported into the endoplasmic reticulum (ER) through the TAP 1 molecule, and loaded on MHC class I molecules in the ER (105). The MHC class I-antigen complex is presented on the DC surface for CD8+ T cell recognition. Alternatively, antigens can be directly degraded in the endosome by phagosomal degradation and loaded on MHC class I molecules (106).
Several previous studies demonstrated that endocytosed CPP-cargo proteins exhibit functions as enzymes, transcription factors, or inhibitors suggesting that CPP-proteins can escape the endosome. For this reason, the CPP-antigen can also be processed for antigen presentation via the same pathways used for naturally occurring antigens (107). The cytosolic delivered antigen can be presented on MHC class I, and they play a critical role in the efficacy of DC vaccinations. Some amount of endocytosed CPP-antigen also can be trapped in the endosome and undergone phagosomal degradation and MHC class II or MHC class I presentation. Because CPPs use the endocytic pathways of dendritic cells, CPP-antigens could efficiently share antigen presentation mechanisms (Fig. 1).
Advantages and limitations of CPP coupling to antigens in DC vaccination
Previous studies demonstrate that CPP-mediated DC vaccination approaches are promising, especially ex vivo DC manipulation, because of the highly enhanced antigen delivery efficiency. It has been shown that R9 linked antigens can induce immune responses with higher efficiency than TAT, suggesting that higher cargo delivery ability leads to higher immune responses (100). In a recent paper, the in vivo immune response boosting ability by CPP-using DC vaccination was much higher than antigen only (101). It is presumably due to CPP linked cargo molecules is preferentially delivered into phagocytic cells compared to other immune cell types (48).
The advantage of cell-penetrating peptides over DC surface molecule specific antibody methods has been reported. A previous study demonstrated that delivery and cross presentation efficiency was not significantly different between a DC-SIGN targeting antibody and the CPP coupling method (108). Using in vivo delivery of CPP-antigen molecules to bypass the expensive and laborious ex vivo DC manipulation method does not overcome the issues of antigen specificity because despite the many advantages of CPPs, they are still regarded as a nonspecific delivery tool (109). Immunogenicity of CPP itself is still concerned as a critical limitation especially if it is originated from non-self while Hph-1-ctCTLA-4 which is human chimeric protein did not induce specific antibody (46). In addition, appropriate CPP-antigen coupling via covalent or non-covalent conjugation and its purification processes is another technical hurdle for versatile application. Nevertheless, the advantages of using CPP for DC vaccination are important for effective therapy and include efficient cytosolic delivery through endosome escape, induction of both CD4+ and CD8+ T cell responses, DC surface molecule independency, and high efficiency.
CONCLUSION
CPPs are considered an attractive therapeutic application tool and numerous approaches utilizing CPPs were reported to successfully treat various diseases in animal models. CPP coupling to antigens in DC vaccination strategies have also recently been highlighted in cancer therapy. CPP-tumor antigens for ex vivo DC manipulation efficiently enable induction of antigen specific effector T cell responses, including CTL responses. The direct application of CPP-antigen for in vivo targeting has also been successful in generating antigen specific immune responses. However, the current limitation of CPP coupling to a limited number of antigens and still less in vivo efficiency requires further investigation. A way of overcoming this limitation would be to use highly efficient CPP for in vivo delivery or various other coupling methods involving simultaneous use of several antigens in order to broaden its application. Based on the understanding of current DC vaccination strategies, we expect to develop successful therapeutic and/or preventive DC vaccinations for treatment of cancer or infectious diseases.
 XML Download
XML Download