

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abbreviations
T1D
type 1 diabetes
TLR
toll-like receptor
DC
dendritic cell
NOD
nonobese diabetic
DPP4
dipeptidyl peptidase
Mφ
macrophage
DAMP
death-associated molecular pattern
APC
antigen-presenting cell
CRAMP
cathelicidin-related antimicrobial peptide
GLP-1
glucagon-like peptide 1
iPSc
induced pleuripotent stem cell
INTRODUCTION
While the last step in the development of type 1 diabetes (T1D) is death of β-cells producing insulin, the initial step or "the initial event" of T1D has been unclear. Recent papers have shown the role of β-cell death in the initial course of T1D, suggesting that β-cell death or apoptosis is α and ω in the pathogenesis of T1D. Such findings provide not only innovative scientific clues to the understanding of T1D but also a novel strategy for the inhibition of treatment T1D. In this review, we will summarize recent findings regarding the relationship between β-cell death and autoimmunity, and their application to the prevention or treatment of T1D.
β-CELL APOPTOSIS AS A MECHANISM OF DIABETOGENIC T CELL SENSITIZATION IN T1D
In T1D, apoptosis of pancreatic β-cells appears to be the last step in the development of diabetes. After a long sequence of autoimmune processes, finally β-cell apoptosis occurs and clinically overt diabetes ensues when β-cell mass is lowered below a certain threshold (1,2). In contrast, the initial event in the development of T1D has been unclear, while the initial event could be more important both scientifically and clinically compared to the last event. Clues to the initial event came after the elucidation of the cell death mechanism and cloning of innate immune receptors. Suggestion that cell death and the initiation of autoimmunity are interrelated was from the observation that there is a transient wave of physiological β-cell apoptosis peaking at 2~3 weeks of age during the pancreas organogenesis (3,4), which coincides with the onset of autoimmunity to β-cell autoantigens in nonobese diabetic (NOD) mice developing classical T1D. Apoptotic cells are rapidly engulfed by macrophages (Mφs), thus no inflammation ensues after apoptosis. In contrast, necrotic cells release death-associated molecular pattern (DAMP) and can stimulate antigen-presenting cells (APCs) such as Mφs or dendritic cells (DCs), which acts as an alarm signal for immune stimulation. Mφs of NOD mice have been reported to have defect in their phagocytic capability, which may allow apoptotic cells to undergo secondary necrosis. Such secondary necrotic β-cells can induce inflammation or immunity through innate immune receptors. Indeed, we observed that insulinoma cells undergoing secondary necrosis can stimulate Mφs in a TLR2-dependent pathway, inducing inflammatory cytokine release from Mφs (5). We also found that and priming of diabetogenic T cells by DCs in vivo that occurs selectively in pancreatic lymph nodes due to β-cell death is also dependent on TLR2. In vivo role of TLR2 in the development of T1D was further supported by a marked decrease in the incidence of diabetes in two types of T1D animal models. multiple low-dose streptozotocin model and spontaneous T1D model of NOD mice (5). Role of TLRs other than TLR2 in T1D has also been suggested in a recent paper reporting that activation of TLR9 of plasmacytoid DCs by a complex of self DNA, anti-double-strand DNA antibody released from B-1a lymphocytes in response to β-cell apoptosis and DNA-binding cathelicidin-related antimicrobial peptide (CRAMP) released from neutrophils in the initiation of T1D (6).
INHIBITION OF T1D BY TLR2 TOLERANCE
These results showing the sensing of DAMP from β-cells by TLR2 on DCs in the initial step of the development of T1D imply that TLR2 blockade could be employed to inhibit autoimmune diabetes. Thus, we employed the strategy of TLR2 tolerance induction which is similar to the well-known LPS tolerance (7). Indeed, when we administered a TLR2 agonist, Pam3CSK4 to NOD mice since 3 weeks of age, the incidence of diabetes was significantly suppressed, suggesting that TLR2 tolerance can inhibit the development of T1D (8). The inhibition of T1D by chronic treatment with Pam3CSK4 could be attributed to TLR2 tolerance of DCs since diabetogenic T cell proliferation in pancreatic lymph nodes by DCs was significantly suppressed after prolonged treatment with Pam3CSK4 (Fig. 1A). Induction of costimulatory signals on DCs by in vitro incubation with Pam3CSK4 was also attenuated by chronic administration of Pam3CSK4 in vivo for 3 weeks, again attesting the induction of DC tolerance (8) (Fig. 1B). On the other hand, the expression of costimulatory molecules on DCs was not changed by in vivo treatment with Pam3CSK4 alone, suggesting that prolonged Pam3CSK4 administration itself does not activate DCs (Fig. 1C). When we studied the molecular mechanism of TLR2 tolerance by chronic treatment with TLR2 agonist, decreased expression of IRAK-1 and -4, positive regulator of TLR signaling, and increased expression of IRAK-M, a negative regulator of TLR signaling were noted. Downregulation of IRAK-1 and -4 protein levels was due to proteasomal degradation as the reduced IRAK-1 and -4 protein levels were restored by proteasomal inhibitors. In this regard, a recent paper showed contribution of IRAK-4 to TLR2 tolerance but not to endotoxin tolerance (9), which can explain the selective TLR2 tolerance after treatment with Pam3CSK4 without heterotolerance to endotoxin (8). Besides DC tolerance, other immune mechanisms such as changes in Th1/Th2 polarization or Treg cells can play a role in the inhibition of T1D after chronic treatment with Pam3CSK4. However, we observed no changes in the Th1/Th2 polarity and the number or activity Treg cells after Pam3CSK4 treatment, while we cannot totally eliminate the role of Th1/Th2 polarization or Treg cells in the inhibition of T1D by Pam3CSK4 (8).
TREATMENT OF ESTABLISHED T1D BY TLR2 TOLERANCE IN CONJUNCTION WITH MEASURES INCREASING β-CELL MASS
While T1D of NOD mice could be inhibited by TLR2 tolerization, immune tolerance alone is not enough for the treatment of established T1D because β-cell mass is already critically reduced below a certain threshold in clinically overt T1D. Immune tolerance in established T1D is also different from that in prediabetic animal models in that immune tolerization should be targeted to sensitized T cells rather than naive T cells. To confirm that TLR2 tolerance could be induced in sensitized T cells, adoptive transfer experiment was conducted in which transfer of sensitized T cells from diabetic NOD mice rapidly induces diabetes in nondiabetic NOD mice. When we administered Pam3CSK4 to recipient mice since just before adoptive transfer of T cells from diabetic mice, the development of diabetes in the recipient mice was significantly suppressed (Fig. 2), suggesting that TLR2 tolerization can block activity of sensitized T cells as well. Sustained interaction between DCs and diabetogenic T cells may be necessary for full in vivo activity of effector T cells. To attain another goal for the treatment of established T1D-replenishment of critically reduced β-cell mass, we employed a well-known strategy of islet transplantation. When islets were grafted under the kidney capsule of newly diabetic NOD mice, blood glucose was normalized immediately. However, diabetes recurred in 10 days because already established autoimmunity destroys islet graft. In contrast, when we administered Pam3CSK4 since just before islet transplantation, blood glucose was maintained below diabetic level in most mice as long as Pam3CSK4 was administered (8), suggesting successful treatment of newly established T1D by a combination of TLR2 tolerance and islet transplantation.
While established T1D could be treated by a combination of TLR2 tolerization and islet transplantation, supply of islets is too limited to cover clinical demand, which preclude the clinical application of islet transplantation for the treatment of established T1D. We thus employed another strategy of increasing reduced β-cell mass. Recently, glucagon-like peptide 1 (GLP-1) mimetics or dipeptidyl peptidase 4 (DPP4) inhibitors that inhibit degradation of GLP-1 by DPP4 in vivo, are being used as new classes of anti-hyperglycemic agents with β-cell trophic action not only in animal model of diabetes (10,11) but also in humans (12). We investigated whether a combination of DPP4 inhibition by DA1229, a novel inhibitor of DPP4 (13) and TLR2 tolerance could treat established T1D. Indeed, euglycemia was achieved in more than 80% of newly diabetic NOD mice after chronic treatment with Pam3CSK4 in combination with DA1229, while treatment with Pam3CSK4 or DA1229 alone was without effect (14). Consistent with the improved blood glucose level, β-cell mass was significantly increased after combined treatment with Pam3CSK4 and DA1229 but not after treatment with Pam3CSK4 or DA1229 alone. The increase in β-cell mass could be attributed to the enhanced proliferation of β-cells as evidenced by increased number of bromodeoxyuridine-incorporating β-cells and an increased number of small β-cell units that comprise less than four β-cells and represent β-cell neogenesis, which was observed in diabetic NOD mice treated with Pam3CSK4 in combination with DA1229 but not in those treated with Pam3CSK4 or DA1229 alone (14). These data suggest the possibility that a combination of TLR2 tolerization and DPP4 inhibition could employed as a new method for the treatment of established T1D without islet transplantation.
CONCLUSION
While it has been elusive for a long time, the real identity of the initial event of T1D is finally emerging, which was owing to the elucidation of the relationship between cell death and innate immune responses and characterization of innate immune receptors. Such a revelation is likely, to lead to the development of novel strategies for the prevention or treatment of T1D. Methods increasing β-cell mass is already available such as DPP4 inhibitors or GLP-1 mimetics. Better methods to increase β-cell mass will be available in the future future since the discovery of inducible pleuripotent stem cell (iPSc) technology (15). By combining such methods of increasing β-cell mass and inhibition of the initial event of T1D, clinically applicable methods of treating established T1D patients will be finally available.
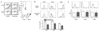

 XML Download
XML Download