

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Glial cells such as microglia and astrocytes play a supportive role in the central nervous system (CNS): for example, microglia form a first-line of defense protecting the CNS from pathogens and other harmful conditions; and astrocytes maintain homeostasis in the CNS microenvironment by regulating neurotransmitter and ion metabolism. In addition to these physiological functions, glial cells also participate in chronic neuroinflammation under pathological conditions. Long-lasting and excessive activation of glia contributes to neural tissue damages in neuroinflammatory and neurodegenerative diseases such as multiple sclerosis, Alzheimer's disease, Parkinson's disease, and HIV-associated dementia (1-3). Cell migration and morphological changes are closely associated with chronic activation of glia. Activated glial cells often show characteristic changes in migratory and morphological phenotypes, which are collectively referred to as reactive gliosis. Molecular mechanisms underlying reactive gliosis has been a subject of intensive investigation.
Lipocalin 2 (LCN2) is a small hydrophobic molecule-binding protein, which is also called 24p3 or neutrophil gelatinase-associated lipocalin (NGAL). LCN2 plays an important role in diverse cellular processes, such as cell death/survival (4-6), cell migration/invasion (7,8), cell differentiation (9,10), iron delivery (4,9,11,12), and insulin resistance (13). Nevertheless, little is known about the role of LCN2 in the CNS. Recently, we have reported that the LCN2 protein upregulates chemokine expression in the CNS (14), and LCN2 promotes morphological changes and cell migration in an autocrine or paracrine manner (15,16). In this study, we focused on the role of soluble mediators secreted by LCN2-activated astrocytes, rather than the direct effects of LCN2 itself. For this purpose, conditioned media obtained from LCN2-stimulated astrocytes were tested for their potential effects on glial and neuronal migration and morphology. In the second half of this study, in vivo role of LCN2 was examined using a zebrafish model.
MATERIALS AND METHODS
Reagents and cells
The recombinant mouse interferon-γ (IFN-γ proteins were purchased from R&D Systems; Minneapolis, MN). The recombinant mouse LCN2 protein was prepared, as previously described (16). In brief, the recombinant mouse LCN2 protein was expressed as a glutathione S-transferase (GST) fusion protein in the BL21 strain of E. coli, which does not synthesize siderophore. The protein was purified by using glutathione-Sepharose 4B beads (GE Healthcare, Princeton, NJ). All other chemicals, unless otherwise stated, were obtained from Sigma Chemical Co. (St. Louis, MO). The mouse primary astrocyte and microglia cultures were prepared from the brains of 0-to 3-day-old ICR mice (Samtako Co., Osan, Korea), as previously described (16). Primary cultures of dissociated cerebral cortical neurons were prepared from embryonic day 20 (E20) ICR mice, as described previously (17,18). The purity of the glial or neuronal cultures was determined by immunocytochemical staining, using antibodies against microglia-, astrocyte-, or neuron-specific markers. Animals used in the current research were acquired and cared for in accordance with guidelines published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The study was approved by the Institutional Review Board of the Kyungpook National University School of Medicine.
Preparation of astrocyte-conditioned media
To prepare astrocyte-conditioned media (ACM), primary astrocytes prepared from the brains of 0~3-day-old ICR mice were cultured at the density of 1.5×106 cells in 100 mm plates in DMEM, supplemented with 10% FBS for 24 hr. Primary astrocyte cultures were treated with the recombinant LCN2 protein (10µg/ml) or left untreated for 24 hr. Cells were then washed twice with PBS, and cultured in fresh DMEM for an additional 24 hr. The ACM was then collected, centrifuged at 1,000 rpm for 5 min to remove cell debris, and stored at -80℃ until further analysis.
In vitro cell migration assays
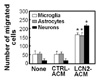
Cell migration was determined by using a 48-well Boyden chamber (NeuroProbe, Gaithersburg, MD), according to the manufacturer's instructions. ACM was placed into base wells separated from the top wells by polyvinylpyrrolidone-free polycarbonate filters (8µm pore size; 25×80 mm; Neuro-Probe). Cells were harvested by trypsinization, resuspended in DMEM, and added to the upper chamber at a density of 1×104 cells/well. Cells were incubated at 37℃ under 5% CO2 for 48 hr. At the end of the incubation, non-migrating cells on the inner side of the membrane were removed with a cotton swab. Migrated cells on the underside of the membrane were fixed with methanol for 10 min and stained with Mayer's Hematoxylin (Dakocytomation, Glostrup, Denmark) for 20 min. Photomicrographs of five random fields were taken (Olympus CK2; Tokyo, Japan), and cells were enumerated to calculate the average number of cells that had migrated. All migrated cells were counted, and the results were presented as the mean±SD of triplicates.
Morphological analysis of neuronal cells
The morphological analysis of neuronal cells was performed by using fluorescence microscopy (Olympus BX50). Cells were blocked with 1% BSA in PBS-Tween 20 for 10 min and incubated in PBS containing 3% BSA and mouse anti-microtubule-associated protein-2 (MAP2) antibody (1:600 dilution; Promega). After two washes in PBS-Tween 20, cells were incubated with anti-mouse IgG-fluorescein isothiocyanate (FITC)-conjugated secondary antibody (BD Biosciences, San Jose, CA). Neuronal processes were quantified as previously described, but with a slight modification (19). In brief, the total number of neuronal process that was longer than one cell body diameter was counted. The number of neuronal process was determined from a minimum of five randomly chosen microscopic fields containing at least 200 cells.
Zebrafish experiments
Zebrafish embryos were collected from pair mating, raised at 28.5℃ in egg water, and staged according to hours post-fertilization (hpf) and morphological criteria as previously described (20). Wild-type AB zebrafish line was used for this study. Plasmids used in this study were constructed by MultiSite Gateway recombination cloning system (Invitrogen). To generate her4-5' entry clone, her4 promoter from the pBS-her4 plasmid (21) was ligated into the multiple-cloning site of p5E-MCS entry vector (22). The lcn2-middle entry clone (16), EGFP-middle entry clone (pME-EGFP), EGFP-3' (p3E-EGFP) and polyA-3' (p3E-polyA) entry clones (22) were used. For the generation of her4:lcn2:egfp construct, LR reaction was performed with her4-5' entry, lcn2-middle entry, and egfp-3' entry clones according to the Gateway LR reaction manual (Invitrogen). For the generation of her4:egfp construct, LR reaction was performed with her4-5' entry, egfp-middle (pME-EGFP), and polyA-3' (p3E-polyA) (22) entry clones. The final plasmids were prepared using the EndoFree plasmid kit (Qiagen, Valencia, CA) and injected into one-cell stage embryos at a concentration of 30 ng/µl in 0.1 M KCl solution containing 0.05% phenol red. For immunocytochemistry, embryos were fixed in AB Fix (4% paraformaldehyde, 8% sucrose, 1× PBS) overnight at 4℃, embedded in 1.5% agarose/30% sucrose, and frozen in 2-methyl butane chilled by immersion in liquid nitrogen. Transverse sections (10µm) were collected by using a cryostat microtome, and sequentially stained with mouse monoclonal antibody against HuC/D (1:20 dilution; Molecular Probes, Eugene, OR) and Alexa Fluor 568-conjugated goat anti-mouse IgG (Molecular Probes). The relative migration of neuronal cells of zebrafish embryos was quantified by comparing the number of neurons located in the medial position with the number of neurons in the lateral margin of the spinal cord. Ten spinal cord sections for each embryo were quantified.
Statistical analysis
All data were presented as mean±SD from three or more independent experiments, unless stated otherwise. Statistical comparisons between different treatments were done by either a Student's t-test or one-way ANOVA with Dunnett's multiple-comparison tests by using the SPSS version 14.0K program (SPSS Inc., Chicago, IL). Differences with a value of p<0.05 were considered to be statistically significant.
RESULTS AND DISCUSSION
We have previously demonstrated that LCN2 upregulates chemokine expression in brain astrocytes (14). These results led us to hypothesize that chemokines secreted by astrocytes may regulate cell migration in the CNS. This hypothesis was tested by evaluating the effect of LCN2-treated astrocyte conditioned media (ACM) on the CNS cell migration. Boyden chamber assay revealed that LCN2-treated ACM (LCN2-ACM) enhanced migration of microglia, astrocytes, and neurons (Fig. 1). It has been also previously reported that LCN2 is an autocrine mediator of reactive astrocytosis (16). Thus, LCN2-upregulated chemokines may also modulate the morphological phenotype of CNS cells. Indeed, LCN2ACM significantly increased the number of process in cortical neurons (Fig. 2). The results indicate that LCN2 regulates CNS cell migration and morphology through secretion of chemokines and other soluble mediators.
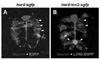
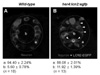
In the next set of experiments, in vivo role of LCN2 was examined in a zebrafish model. For the ectopic expression of lcn2 in the zebrafish CNS precursor cells, we first generated her4:lcn2:egfp and her4:egfp DNA constructs, which express LCN2-EGFP fusion protein and EGFP alone, respectively, under the control of her4 promoter (21). Each DNA construct was injected into zebrafish embryos at the one-cell stage. Injected embryos were fixed at the 24 hr post fertilization (hpf) and labeled with anti-Hu antibody, which is a marker for neurons in zebrafish CNS (23,24). In the spinal cord of her4:egfp DNA-injected control embryo, EGFP fluorescence was detected in the Hu-non-neuronal precursor cells; Hu+ neurons were normally located in the lateral margin of the spinal cord at the 24 hpf (Fig. 3A). However, her4:lcn2:egfp DNA-injected embryos showed abnormal localization of neurons; neuronal cells near the LCN2:EGFP-expressing cells were located in the more medial position close to the LCN2:EGFP+ cells compared to the control embryo (Fig. 3B), indicating that LCN2 expression attracts neurons. LCN2 expression exerted a similar chemotactic effect, when the number of Hu+ neurons was counted in different regions of the spinal cord, medial position versus lateral margin: percentage of neuronal cells in the medial position of her4:egfp DNA-injected control embryo, 5.60±0.78%; percentage of neuronal cells in the medial position of her4:lcn2:egfp DNA-injected embryos, 11.92±1.39%; the results are mean±SD (n=10; p<0.05) (Fig. 4). These results support that LCN2 regulates CNS cell migration in vivo.
In this study, we present evidence that LCN2 regulates CNS cell migration and morphology possibly by upregulating the secretion of chemokines or other soluble mediators. We have previously reported that CXCL10 is one of the chemokines produced by astrocytes, and CXCL10 induces CNS cell migration and morphological changes. This was demonstrated mostly using cultured glial cells and neurons. In this study, we extended our previous findings to further evaluate the effects of glia-derived mediators on CNS cell migration and morphology. Moreover, zebrafish was utilized as an animal model to confirm the results in vivo. Our findings both in vitro and in vivo settings commonly support that LCN2 modulates CNS cell migration and morphology. These results may deepen our understanding of cellular behaviors of brain glial cells and the molecular mechanisms of reactive gliosis.

 XML Download
XML Download