

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Transforming growth factor-β (TGF-β) superfamily consists of a large family of structurally related cytokines, which are classified into two main groups: TGF-β/Activin and bone morphogenetic proteins (BMPs) and growth/differentiation factors (GDFs) (1).
Among them, TGF-β has been well characterized as a representative potent immunosuppressive cytokine. TGF-β has been reported to inhibit immune system by modulating both effectors and suppressors. TGF-β directly inhibits innate immune cells such as macrophages and NK cells and adoptive immune cells such as CD4+ helper T cells and CD8+ cytotoxic T cells, while expanding immunosuppressive regulatory T cells (Tregs) and inducing the recruitment of immunosuppressive myeloid-derived suppressor cells (2).
The immune surveillance system is one of the most important defense mechanisms against cancer progression. Attempts to enhance tumor immunity by various strategies have been tried as supportive anti-cancer therapies. The most potent immunosuppressive cytokine, TGF-β is massively produced and activated in tumor microenvironment, which allows invasive metastatic cancers to escape from immune surveillance (3). Thus, antagonizing TGF-β's functions has been implicated as the most potent anti-tumor immune therapy and some TGF-β antagonists have been on preclinical and clinical trials (4-6).
PROGNOSIS AND THE SIZE/NUMBER OF METASTASIS
Tumor recurrence by remote metastasis affects the prognosis of cancer patients after radical surgery. Cancer staging is evaluated by TNM classification, which describes the size and the extent of the Tumor, degree of the spread to regional lymph Nodes and the presence of Metastasis. TNM staging is widely employed as a global standard to plan the treatments and to indicate the prognosis (7). Although there are no detailed parameters in metastasis defined in TNM staging, the size and the number of metastasis significantly affect the prognosis of the patients. In pulmonary metastasis from breast cancer, it has been reported that the size and the number of metastasis significantly correlate with prognosis and re-recurrence and the pulmonary metastasectomy is justified in case of small number of metastasis lesions (8). It has been reported that an increase in tumor size and an increase in number of regional lymph nodes with metastasis were associated with diminution in cellular immunity (9), which might be the consequence of the production of TGF-β by tumors themselves and tumor infiltrating immunosuppressive cells (10,11). Thus, one might expect that the number and the size of tumor and metastasis could be reduced by blocking TGF-β to achieve better prognosis.
TGF-β AND TUMOR IMMUNE SURVEILANCE-AFFECTING NUMBER OF METASTASIS
Although some tumors express tumor specific antigens, which can be recognized by immune system, tumor specific immune responses often fail to eradicate tumors because tumors evade immune surveillance by variety of strategies. Tumors themselves, tumor stromal cells and the myeloid-derived suppressor cells produce large amount of TGF-β, which is activated in the tumor microenvironment (12-14). By its potent immunosuppressive effects, TGF-β strongly inhibits anti-tumor immunity.
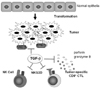
Partly due to TGF-β, loss or down-regulation of MHC class I molecules is frequently observed in malignant cells, which makes them invisible to immune system (15). Poorly immunogenic tumors with low expression of MHC class I can escape from attack by cytotoxic CD8+ T cells but they become more sensitive to attack by NK cells (16). TGF-β also affects the tumor cell-NK cell interaction by modulating the expression of MHC class I homologues on the tumor cells and their receptors on NK cells. Expression of MHC class I homologues such as MHC class-I-chain-related protein A (MICA) and Rae-1γ human and murine NKG2D ligand, respectively, as well as NKG2D are reported to be down-regulated by TGF-β (17-19) Nam JS et al. demonstrated that anti-TGF-β antibody treatment up-regulated NKG2D expression on CD8+ T cells, not NK cells, which were already highly activated even without anti-TGF-β antibody treatment (19). Anti-TGF-β antibody treatment exerts anti-tumor efficacy by enhancing tumor-immune cell interaction (Fig. 1).
TGF-β directly suppresses functions, migration and expansion of cytotoxic CD8+ T cells, NK cells, NK T cells and γδ T cells, while inducing Foxp3+ Tregs (2). In poorly immunogenic syngeneic 4T1 mammary tumor metastasis model, TGF-β does not significantly affect the infiltration of NK cells and Foxp3+ Tregs, whereas depletion of CD8+ T cells abolished the effect of anti-TGF-β antibody treatment. Treatment with anti-TGF-β antibody increases the infiltration of activated CD8+ T cells expressing CD122, cytotoxic effector molecules such as perforin and granzyme B and Fas ligand in metastatic lesions (19,20).
It is remarkable that CD8+ T cells mainly impact the size of tumor metastasis, whereas NK cells dramatically impact metastasis number in poorly immunogenic 4T1 mammary tumor metastasis model. CD8+ T cells do not significantly affect the number of metastasis. Treatment with anti-TGF-β antibody restores the cytolytic CD8+ T cell activity similar to NK cell activity to reduce tumor metastasis number to some extent, although very highly activated NK cells are not significantly affected by anti-TGF-β antibody (19). When TGF-β antagonists are administered systemically, untoward effects by autoimmune reactions might be possible (2). However, anti-TGF-β antibody increases cytotoxic T cell activity only locally at the tumor sites without activating systemic immune system, which indicates that the clinical application of TGF-β antagonists to cancer patients is tolerable (4-6,19,20).
TGF-β AND TUMOR IMMUNE SUBVERSION-AFFECTING SIZE OF TUMOR AND METASTASIS
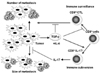
The balance between the effector cell populations and the suppressor cell populations finely tunes immune system. In malignant tumor environment, the suppressor populations such as Foxp3+ Tregs and suppressor myeloid cells dominate over effector cell populations and suppress the effective anti-tumor immunity. Large amount of TGF-β produced and activated in tumor environment promotes the development and migration of these suppressor populations (21,22).
Recently, the novel subset of CD8+ T cells is identified, which promotes growth of primary and metastatic tumor (23). This CD8+ T cell subset is distinct from the reported CD8+ suppressor/regulatory T cells (24). IL-17 is produced by this subset, which favors tumor growth by preventing tumor apoptosis. The same combination of cytokines to induce Th17 CD4+ T cells in vitro, TGF-β and IL-6, subverts CD8+ T cells into this IL-17 producing CD8+ T cells (Fig. 2).
The importance of IL-17 in inflammatory autoimmune disorders was first addressed by Nakae S et al. (25) and the CD4+ T cell subset producing IL-17 was defined as Th17 by Stockinger B et al. (26). Although T cells infiltrated in tumors produce IL-17 (27), it has been controversial whether IL-17 promotes or inhibits tumor growth (28,29). Recently, it is reported that IL-17 produced by CD4+ T cells, not CD8+ T cells, promotes primary tumor growth through IL-6-Stat3 signaling pathway (30).
In poorly immunogenic 4T1 metastatic mammary tumor model, CD8+ T cell depletion decreases the primary tumor volume as well as metastasis size, indicating that some CD8+ T cells are not cytotoxic, but on the contrary, favor the tumor growth (23). IL-17 produced by tumor infiltrating CD8+ T cells was identified as the major soluble factor to prevent tumor cell apoptosis, and the knockdown of IL-17 response of tumor cells reduces tumor growth by enhancing apoptosis in vivo. IL-17 alone or in combination with TGF-β suppresses apoptosis of tumor cells under the condition with nutrient deprivation or the treatment with apoptosis inducer. It might not be universal phenomena for all the malignant tumors, because some tumor cell lines do not respond to IL-17 and in some tumors, IL-17 oppositely induces apoptosis. However, some patients exhibiting the up-regulation of IL-17 expression in tumor (23,27) might benefit from anti-TGF-β treatment through the same mechanism.
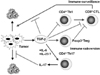
It has been reported that IL-23 is required for autoimmune inflammation mediated by Th17 cells in vivo. It is consistent with the results of lower numbers of IL-17+ T cells in IL-23 deficient mice. These findings suggest that IL-23-dependent signaling in Th-17 cells may depend on the transcription factor Stat3 (31).
Similarly, it is recently reported that Stat3 promotes IL-23-mediated procarcinogenic immune response while inhibiting IL-12-dependent antitumor immunity (32). However, it is not yet determined whether IL-23 is required for the development of IL-17-producing CD4+ T cells and IL-17-producing CD8+ T cells infiltrated in tumor in vivo. They also report that Stat3 is phosphorylated through IL-23 receptor to up-regulate the expression of Foxp3 and immunosuppressive cytokine, IL-10 in CD4+ T cells (Fig. 3). It is also to be determined whether TGF-β is involved in this effect. The IL-17/IL-23 axis might be the new mechanism of tumor immune subversion (33).
PERSPECTIVE
Researchers used to focus on tumor immune surveillance by effector cell populations to enhance tumor immunity. Recent accumulation of data shows the importance of regulating tumor immune subversion by suppressor cell populations to reverse immune suppression against tumor. Antagonizing TGF-β successfully enhances tumor immune surveillance and reverses immune subversion, which results in the decrease in the number of metastasis and the size of tumor and metastasis. So far, TGF-β antagonists are applied to the treatment of terminal stage patients of certain highly malignant cancers only in clinical trials. By further confirmation of their efficacy and little toxicity, their application might be expanded to the patients with general cancers before the radical surgery to decrease the size of primary tumors and to prevent metastasis or to the patients before the metastasectomy to reduce the size and the number of metastasis.
 XML Download
XML Download