

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Dilated cardiomyopathy (DCM) is characterized by left ventricular systolic dysfunction and is the most common cause of heart failure in young adults. DCM can result in a large medical cost burden because of repeated hospitalizations and the potential need for transplantation. The prevalence of DCM is greater than 1 in 2500 individuals in the general population, however the prevalence in Korea has not yet been elucidated.1) According to the Korean Heart Failure registry, cardiomyopathy is one of the major underlying causes of acute heart failure.2)
Idiopathic DCM can be diagnosed after exclusion of identifiable non-genetic causes and hereditary cardiomyopathy is a major cause of idiopathic DCM. The prevalence of the familial form is about 25-35%, and using recent advances in genetic diagnostics up to 50% of idiopathic DCM patients were determined to have hereditary cardiomyopathy.3) Currently, more than 40 genes have been identified as causative gene associated with hereditary DCM. Those genes predominantly encode two major cardiac proteins: sarcomere and cytoskeletal. Inherited DCM is a known monogenic disorder which is primarily transmitted as an autosomal dominant trait, although autosomal recessive, X-linked, or mitochondrial inheritance patterns were also identified.
A genetic diagnosis can confirm a hereditary DCM clinical diagnosis and it can help predict disease risks for family members before the onset of symptoms. However, the clinical applications are limited, due to low detection rate and high cost. Since 2013, the Korean government has supported genetic diagnoses for rare genetic disorders, including genetic cardiomyopathy and arrhythmia (http://helpline.nih.go.kr). To diagnose idiopathic DCM, we applied a next generation sequencing (NGS) based multi-gene panel testing and subsequent confirmation with traditional Sanger sequencing analysis.
This review focused on recent advances in genetic diagnoses for idiopathic DCM, characterization of gene mutations in Korean patients, and clinical applications.
Major Genes that Cause Hereditary Dilated Cardiomyopathy
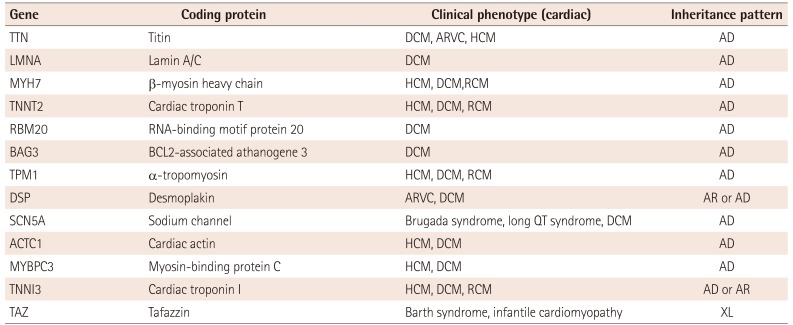
Over 40 genes have been identified as DCM causative genes, and a large proportion of these genes encode for sarcomeres and cytoskeletal elements. Major DCM-causing genes are shown in Table 1, and many genes cause both hypertrophic cardiomyopathy (HCM) and DCM. Although autosomal dominant inheritance is most common, autosomal recessive and X-linked inheritance have also been reported. However, a low detection rate has made genetically diagnosing DCM challenging in clinical practice. Several years ago, Herman et al.4) reported high prevalence of pathogenic truncating variations in the titin (TTN) gene from patients with idiopathic DCM (in 25% of familial cases and in 18% of sporadic cases), and inclusion of the TTN gene together with recent technological advancements has increased the detection rate by up to 50%. Gardia-Pavia et al.5) reported that genetic mutations were identified in 30-40% of sporadic cases and 70-80% of familial cases of DCM were referred for clinical genetic testing. Mutations in the TTN gene are most frequent (15-25%), followed by lamin A/C (LMNA, 4-8%), b-myosin heavy chain (MYH7, 4-8%), cardiac troponin T (TNNT2, 3-6%), and RNA-binding motif protein 20 (RBM20, 3-6%).
TTN is a large muscle protein that is expressed in both cardiac and skeletal muscle, and plays an important role in sarcomere assembly. TTN provides an extendable scaffold for the contractile machinery and is crucial for myofibrillar elasticity and integrity.6) TTN has 363 coding exons with 1 non-coding exon and undergoes alternative splicing. The mutation that disrupts the structure of full-length titin causes DCM. Genetic evaluation of the entire TTN gene was not previously possible, due to its large size and complexity. However, NGS has made sequencing possible, and more than 100 variations in TTN have been associated with cardiomyopathy; DCM is the most widely represented, followed by arrhythmogenic right ventricular cardiomyopathy and HCM. Nonsense mutations, frameshifts, and splice-site variations were common as well as missense mutations.7) This TTN mutation showed a penetrance over 95% in subjects who were more than 40 years of age, and men with this TTN mutation had adverse events at an earlier age than women.4) However, TTN truncation variation was also detected in control subjects that did not manifest cardiomyopathy (3%), suggesting clinicians should be careful when interpreting TTN variants in individuals without cardiomyopathy.4)
Mutations in LMNA, which encodes type V intermediate filament protein, are also a common cause of DCM. Cardiomyopathy caused by the LMNA mutation usually presents in early to mid-adulthood, and the penetrance rate was estimated as 100% after 60 years of age.8) Conduction system diseases precede heart failure development, which may cause sudden cardiac death with fetal ventricular arrhythmia. About 70% of patients developed a cardiac adverse event within 5 years after diagnosis and it is recommended that patients with these symptoms should consider ICD implantation and/or heart transplantation.9) Family screening can help provide an early diagnosis and may improve outcomes, with proper management.
Mutations in the MYH7 gene, which encodes the b-myosin heavy chain, are very common in patients with HCM (-15% of identified mutations).10) Mutations were also reported in up to 7% of DCM patients, and more than 15 missense mutations have been identified as a cause of DCM.11) HCM may progress to DCM, but there are patients with MYH7 mutations that have not experienced HCM before developing DCM.12)
Mutations in the cardiac troponin T gene (TNNT2) can also cause both hypertrophic and dilated cardiomyopathy.10) Patients with HCM and TNNT2 mutations have malignant prognoses, although the cardiac hypertrophy can be moderate or not significant.13) Several mutations that decrease Ca2+ sensitivity have been associated with DCM, and an animal study showed that Ca2+ sensitizers reduced heart size and improved life expectancy, suggesting that Ca2+ sensitizers may be beneficial in DCM presenting with TNNT2 mutations.10)
RBM20 was first identified by Brauch et al.14) as a causative gene and most mutations were clustered in exon 9. The RNA binding motif protein encoded by RBM20 regulates the splicing of more than 30 proteins including titin,15) and loss of function mutations result in altered expression of proteins that maintain sarcomere structure, leading to cardiomyopathy, fibrosis, arrhythmia and sudden death.15) RBM20 mutations are associated with early onset DCM, end-stage heart failure, and high mortality.
BCL2-associated athanogene 3 (BAG3) was identified as causative DCM gene by Norton et al.16) using whole-exome sequencing and genome wide analysis of copy number variation. The BAG family molecular chaperon regulator 3, encoded by BAG3, is a co-chaperon protein with anti-apoptotic function. Mice with homozygous disruption of BAG3 showed fulminant myopathy characterized by myofibrillar degeneration with apoptosis.17)
Mutations in α-tropomyosin (TPM1), a thin filament sarcomeric gene showed altered electrostatic interactions between actin and tropomyosin.18) The TPM1 gene was reported as a rare cause of HCM, accounting for approximately 3% of cases.13) These mutations, like those in the cardiac troponin T gene, are characterized by relatively mild or subclinical hypertrophy but a high incidence of sudden death. Using a candidate gene approach, Olson et al.18) identified two heterozygous missense mutations of the TPM1 gene in two familial cases with DCM.
Desmosomes are a molecular complex of cell adhesion proteins and are important for maintaining rigidity and cell strength. Desmoplakin, encoded by DSP, is the most abundant among desmosomes. Norgett et al.19) reported the first recessive human mutation in the DSP gene that causes a generalized striate keratoderma, woolly hair, and dilated left ventricular cardiomyopathy. A number of patients with this syndromic disorder developed heart failure in their teenage years, resulting in early morbidity. Mutation in DSP was also identified in arrhythmogenic right ventricular dysplasia.20)
Mutations in sodium channel protein type 5 subunit α (SCN5A) have been implicated in inherited arrhythmia syndromes, such as long QT syndrome, Brugada syndrome, and ventricular tachycardia.21) Mutations in SCN5A were detected in 1.7% of probands in DCM cohort families, and most carriers manifested variable arrhythmia, supraventricular arrhythmia, ventricular tachycardia, and conduction disease. Therefore, based on this information, a DCM patient with a SCN5A mutation should be evaluated for arrhythmia.
Mutations in cardiac actin (ACTC1) were identified in hereditary DCM. However, no mutations were identified in Japanese patients with DCM, suggesting that a mutation in the ACTC1 gene is a rare cause of dilated cardiomyopathy, at least in Japanese patients.22) In addition, ACTC1 mutations were not detected in our population.
Mutation in the cardiac myosin-binding protein C (MYBPC3) is also a common cause of HCM. However, it was reported as a cause of DCM with variable clinical features. HCM patients with MYBPC3 mutations had later onset, lower penetrance, and a better prognosis, compared with patients with mutations in MYH7 or TNNT2.23)
DCM with cardiac troponin I (TNNI3) mutations showed severe, early-onset DCM. Functional analysis revealed that troponin reconstituted with a mutant had lower maximum ATPase rates and reduced Ca2+ sensitivity compared to the wild type.24) TNNI3 gene mutations were also identified in HCM and restrictive cardiomyopathy.
The tafazzin gene (TAZ), which is located on Xq28, is a causative gene for Barth syndrome. Barth syndrome is a rare, X-linked genetic disease characterized by dilated cardiomyopathy with endocardial fibroelastosis, proximal myopathy, growth retardation, neutropenia, organic aciduria, and, particularly, 3-methylglutaconic acid excess.25) Cardiomyopathy is the most common clinical feature in patients with a TAZ mutation, and symptoms usually present early in life.26) TAZ mutations were also identified in infantile cardiomyopathy that affected males, even in the absence of typical clinical Barth syndrome features.
Many genes were not described above because of relatively low frequency in idiopathic DCM patients. Interestingly, many genes are causative of both HCM and DCM, suggesting that substitution of different amino acid residues in the same proteins can impact distinct phenotypes.
Characteristics of Genetic Mutation in Korea
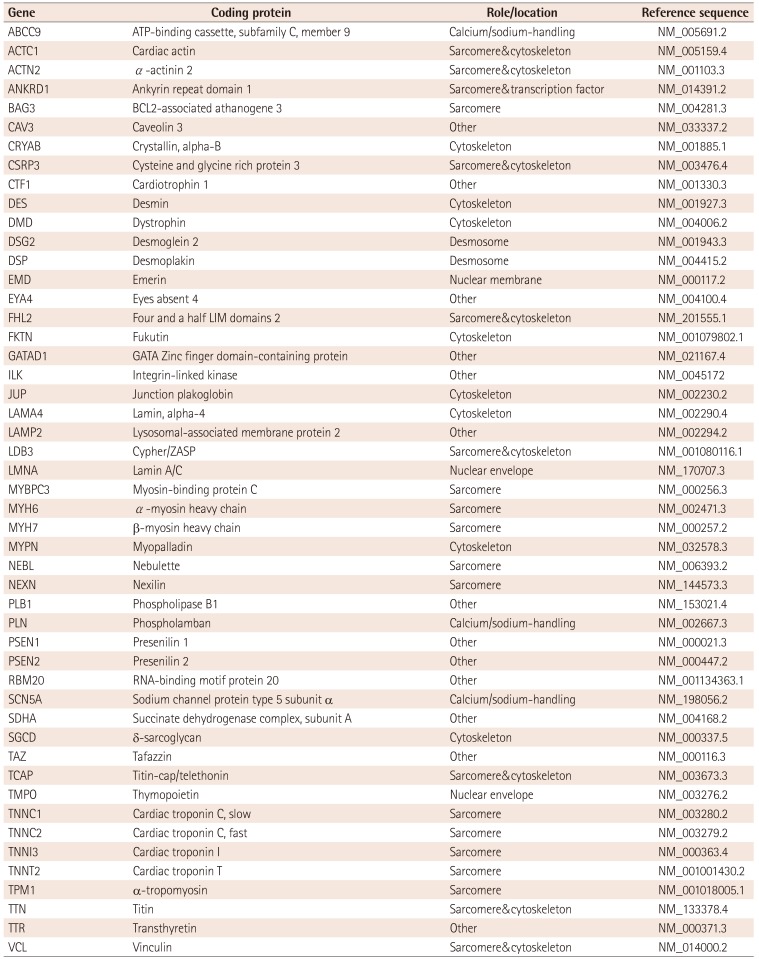
Reports on genetic variations in Korean patients with DCM are limited. The Korea National Institute of Health launched a genetic diagnosis support program for rare diseases in 2013. The Seoul National University and Yonsei University Hospitals cooperatively operate this genetic diagnosis program. Twenty-eight rare diseases were subjected to this support program, and both dilated and hypertrophic cardiomyopathies were included. We applied a targeted gene panel using next-generation sequencing technology with conformation of mutations and Sanger sequencing. For DCM, our test panel was increased from 38 genes in 2013 to 49 DCM genes (Table 2). To date, 88 patients have completed the genetic studies for DCM, and 35 patients (39.8%) were identified with mutations (unpublished data). Nine novel mutations in TTN, MYH6, MYH7, TNNI3, ABCC9, TNNT2 and TAZ were identified as well as 25 known mutations. One whole-gene deletion of LAMP2 was suspected to be causative in subjects with DCM. As expected, the mutation was most frequently found in sarcomere and cytoskeletal genes. Thirty-seven patients (42%) had at least one or more unclassified variants in candidate genes. Sixteen subjects with DCM had no mutation or variants in candidate genes, suggesting the possibility of another candidate gene.
Mutations were commonly identified in MYBPC3, LMNA, and MYH7. We identified MYBP3 mutations at a higher frequency (6 patients, 6.8%) than would be expected based on the known prevalence of DCM.5) Mutations in MYBPC3 accounted for 1% in a previous study, and all mutations were previously identified in HCM patients; therefore we cannot exclude the possibility that there was inappropriate inclusion of patients with end-stage HCM with LV dilatation and systolic dysfunction rather than primary DCM. Further clinical evaluations for these patients are needed. The frequency of LNMA and MYH7 mutations were 5.7% and most of them were previously reported as mutations that caused DCM.
After adding TTN to the gene panel for DCM in 2015, 3 patients were shown to have mutations in the TTN gene, and TTN contributed to about 5% of DCM patients that were referred after addition of TTN to the gene panel. However, this percentage was relatively lower than the detection rate reported by Herman et al.4) TAZ mutations were identified only in children. Our results indicate that targeted gene sequencing is a feasible approach to identify pathogenic mutations in DCM patients. We also identified several novel variants of unknown significance (VUS) in TTN with frequencies less than 1% in control subjects. The clinical significance of these variations should be studied in the future.
Our results were from patients who were referred from cardiology clinics and diagnosed with idiopathic DCM; therefore, the clinical information was limited. In addition, only 88 patients with DCM were analyzed, and this was a small sample size. To further assess prevalence and phenotype characterization, a family-based cohort of DCM patients with a larger sample size is needed. Further studies on the mechanisms of novel mutations are also necessary to elucidate the disease mechanisms.
Genetic Testing in Clinical Practice
Previously, genetic testing of DCM was not easily accessible in cardiology clinics due to genetic heterogeneity and complexity. More than 40 different genes have been identified as causative agents for DCM, and each of these genes provides a modest contribution to the overall incidence of disease. Furthermore, even if genetic testing identifies variants which are not reported in control subjects, it is very difficult to determine whether the mutation is pathogenic.
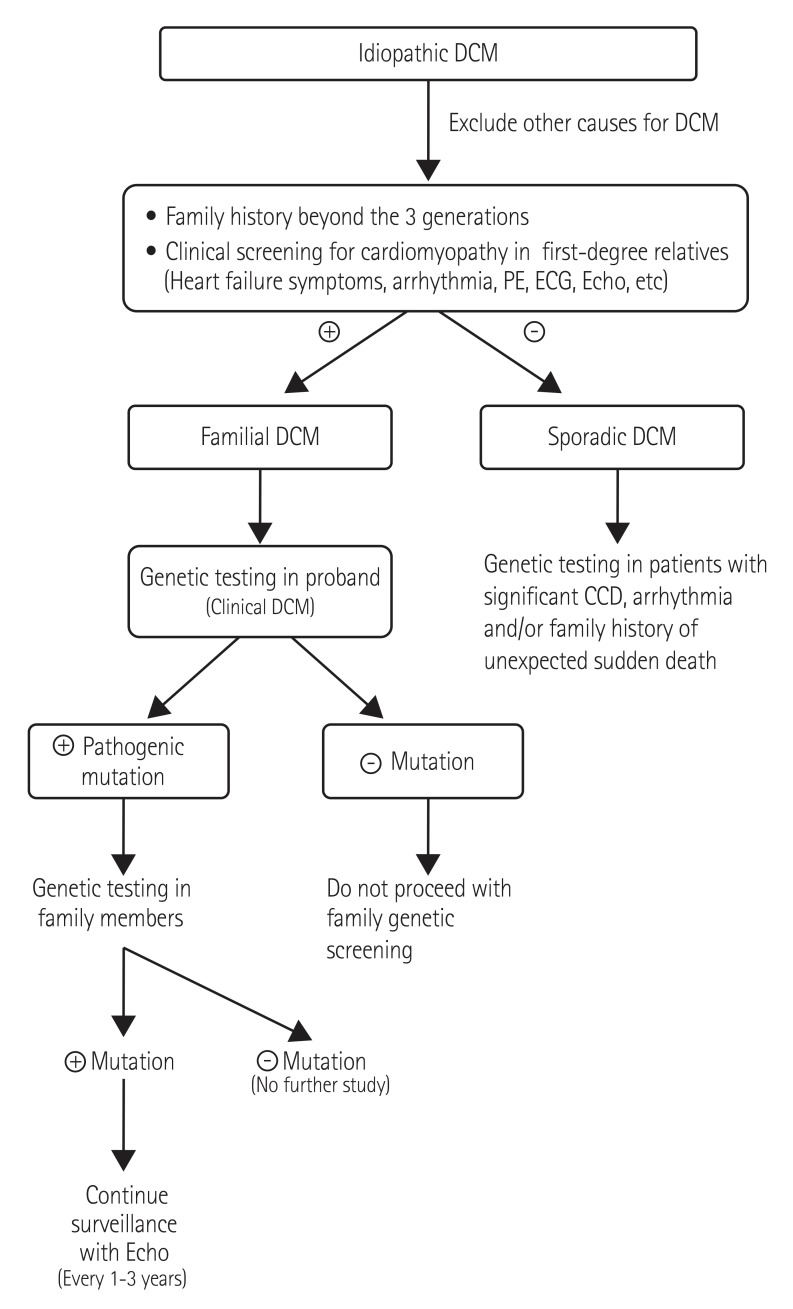
Several guidelines have been developed to aid clinicians in selecting appropriate genetic testing for cardiomyopathies. In 2009, the Heart Failure Society of America published clinical guidelines for genetic evaluation of cardiomyopathy.27) According to these guidelines, a careful family history for more than 3 generations is recommended for all patients with cardiomyopathy, as this may help identify whether their cardiomyopathy is inherited or a transmission pattern. Obtaining a family history is time consuming and it can be difficult for clinicians to obtain sufficient information. Referral to a special clinic with expertise in genetics may be helpful for obtaining and reviewing family history and pedigree information as well as genetic counseling.
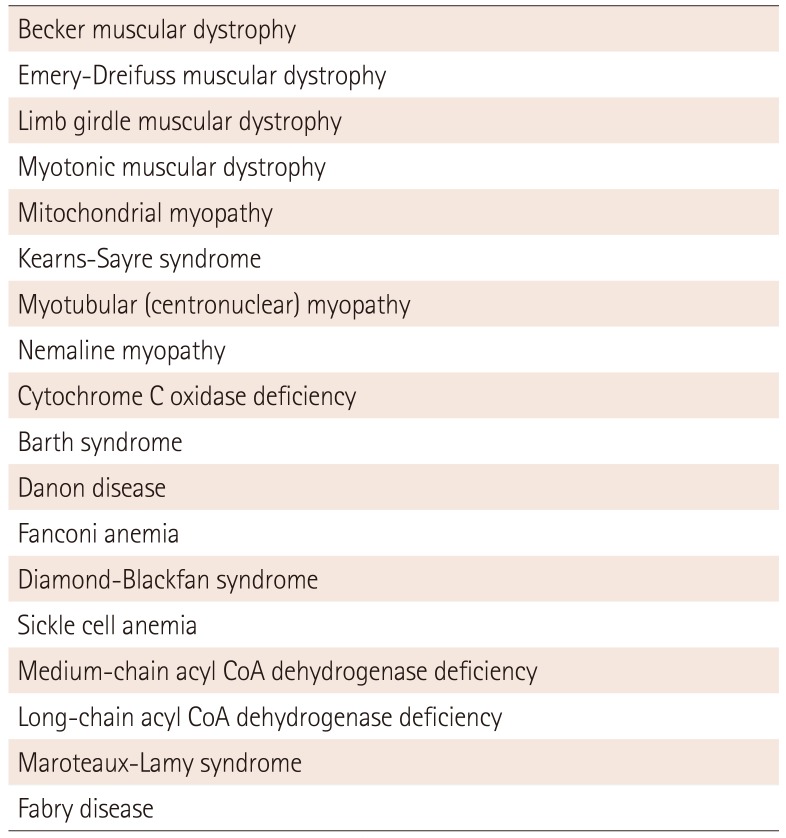
With family history, clinical screening for cardiomyopathy is recommended in asymptomatic first-degree relatives. The clinical screening consists of history taking with special attention to heart failure symptoms, physical examination with special attention to the cardiac and skeletal muscle systems, an electrocardiogram, echocardiogram and creatine kinase isoenzymes (CK-MM). The systemic diseases which should be evaluated and genetic disorders, which can help identify DCM, are listed in Table 3.27)
These guidelines were quite restrictive with respect to molecular genetic testing recommendations. According to the Heart Rhythm Society and the European Heart Rhythm Association (HRS/EHRA) Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies,28) class I recommendation (“is recommended”) for genetic testing for DCM indicated that: (a) comprehensive or targeted (LMNA and SCN5A) DCM genetic testing is recommended for patients with DCM and significant cardiac conduction disease (i.e., first-, second-, or third-degree heart block) and/or a family history of premature unexpected sudden death, (b) mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of a DCM-causative mutation in the index case. A class IIa recommendation (“can be helpful”) included genetic testing for patients with familial DCM to confirm the diagnosis, to recognize those who are at highest risk of arrhythmia and syndromic features, to facilitate cascade screening within the family, and to help with family planning.
However, previous guidelines should be updated because novel technologies have allowed genetic diagnoses to simultaneously test large numbers of genes. Whole-exome sequencing can now complete the sequencing of an entire human genome in several days. Clinical sensitivity, the likelihood that the genetic testing will identify a causative mutation, is an important consideration for genetic testing utilization. The sensitivity of genetic testing has been improved by discovering additional DCM genes, especially TTN. Therefore, genetic testing should be considered in idiopathic DCM.
When a DCM mutation was identified, the clinical screening interval for cardiomyopathy was recommended as annually throughout childhood, every 1-3 years in adults, and at any time that signs or symptoms appeared. If the disease-causing mutation had not been identified, less frequent screening (every 3-5 years, beginning in childhood) was recommended.27)
Modified genetic screening strategies in DCM are shown in Fig. 1. Genetic testing in familial DCM should be considered because identification of mutations helps with early detection in relatives for family planning because clinical sensitivity is more than 70%.5) However the role of genetic testing for sporadic DCM remains uncertain. The clinical effectiveness of gene testing should be re-evaluated in sporadic cases due to technical advances in genetics. Genetic testing interpretation is very important because the detection rate for rare or novel sequence variants is rapidly increasing. In our population, more than 40% of idiopathic DCM patients had rare or novel unclassified variants which should be identified In our population more than 40% of idiopathic DCM patients had rare or novel VUS. Sometimes familial genetic testing can be helpful to determine the clinical significance of VUS in sporadic cases. If a variant is detected in a candidate gene and is not present in parental samples, this variant could be considered evidence of pathogenicity. Therefore clinicians should be careful and educated when interpreting the results of genetic testing. Also, the diagnostic laboratory should provide information to help determine the clinical significance of genetic variants.
Genetic counseling is a complex process that can provide guidance for patients or relatives at risk for genetic disorders and provide them with information, including genetic risk, consequences of the disorder in question, the probability for transmission, disease management and family planning. Genetic counseling should be performed by a genetic counselor, or by a geneticist who is familiar with cardiovascular diseases, or by a cardiologist who is an expert in genetic cardiomyopathy and fluent in genetic counseling. Currently, only a few genetic counselors are available in Korea, therefore cardiologists that identify patients with cardiomyopathy in practice, should be trained for genetic counseling.
Conclusion
During the last decade, technical advances have changed the approaches for genetic diagnoses in many diseases. More than 40 genes have been identified and associated with hereditary DCM. Although we applied a targeted gene panel using NGS technology, which consisted of most known candidate genes, the detection rate was less than 50%. MYBP3, LNMA, and MYH7 were the most frequently identified genes, and the pattern of causative genes was different from previous reports. Developing a more comprehensive test panel with additional DCM genes and using whole exome sequencing will improve the detection rate, and genetic testing can be an option for all patients with idiopathic DCM in near future.
Interpretation of genetic screening tests for DCM remains challenging in clinical practice. Cardiologists should be aware of the limitations of genetic testing. All genetic variations are not pathogenic mutations, and the majority of reported mutations in DCM are unique to a single family, making interpretation of genetic results more difficult. Therefore, integration with clinical features and familial history are needed for clinical diagnoses. Referral to a specialty clinic with expertise in genetics may be helpful for obtaining and reviewing family history and pedigree information, as well as genetic counseling.
 XML Download
XML Download