

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
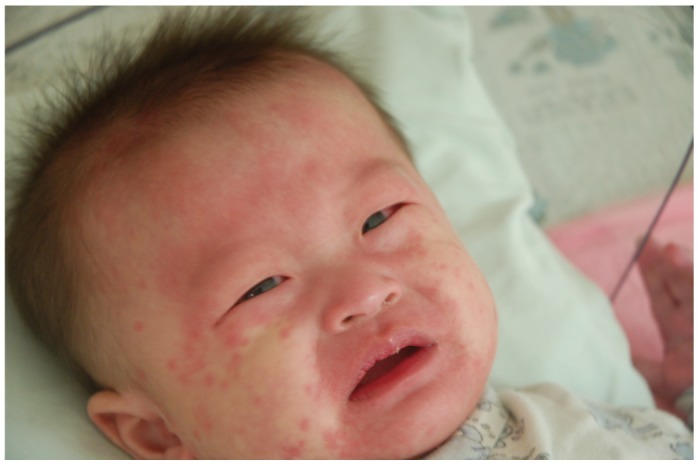
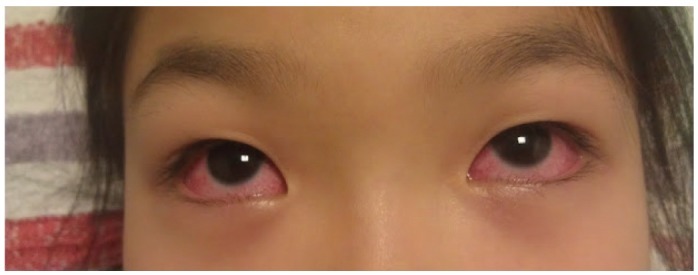
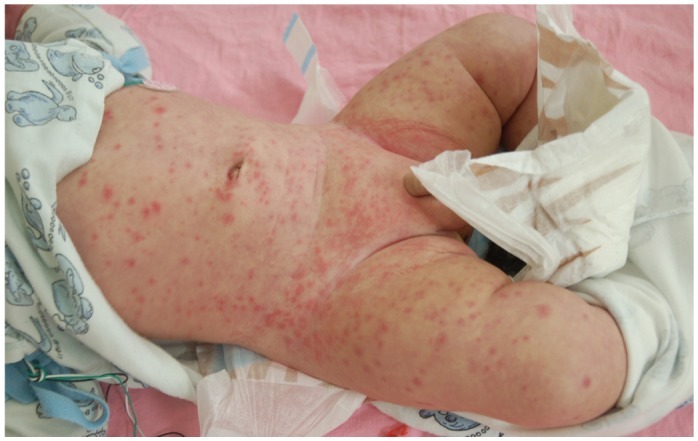
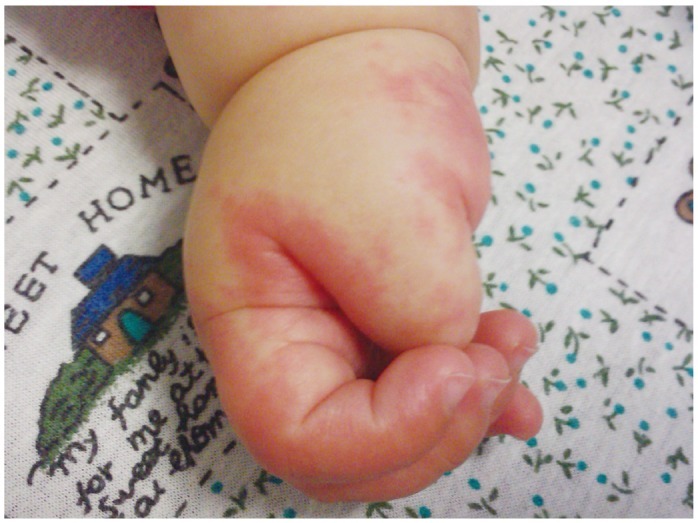
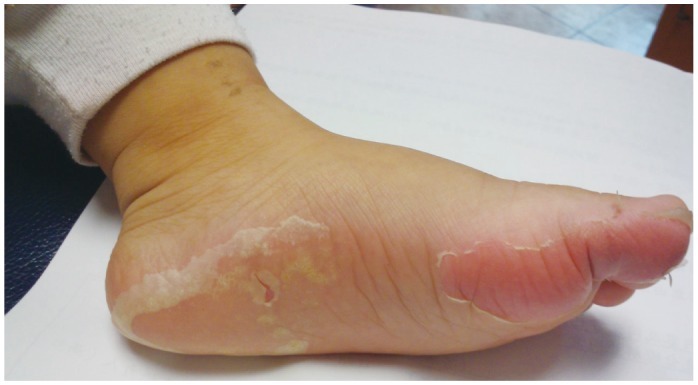
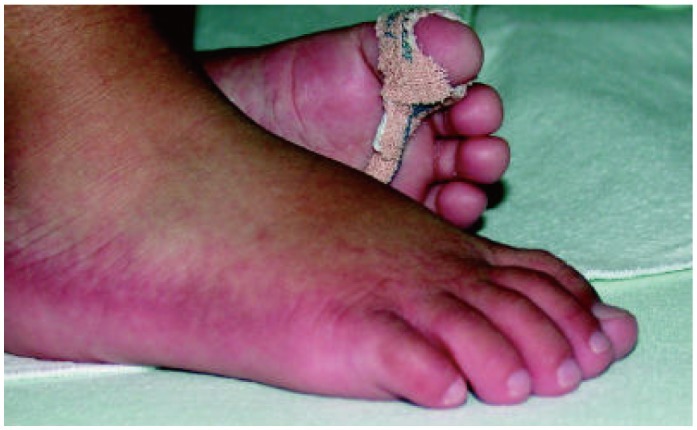
Kawasaki syndrome (KS) is an acute febrile illness which predominately occurs in young children under 5 years of age (75-80%) while it is exceptional in adults (<1%). The clinical picture consists of a persistent, erratically spiking-high fever ranging from 38° to 40℃ (101° to 104°F) which is resistant to antipyretics and antibiotics. In addition to the fever, four out of five of the following principle features are required for diagnosis: 1) a polymorphous rash; 2) conjunctival injection; 3) bright red, swollen extremities with subsequent desquamation typically during the second or third week; 4) oral changes which include bright red fissured lips, oropharyngitis, strawberry tongue; 5) cervical lymphadenopathy (Figs. 1-5). Rash is often the first symptom and appears on the trunk or perineum, which typically spreads around the body. Occasionally, fever is absent in some patients.
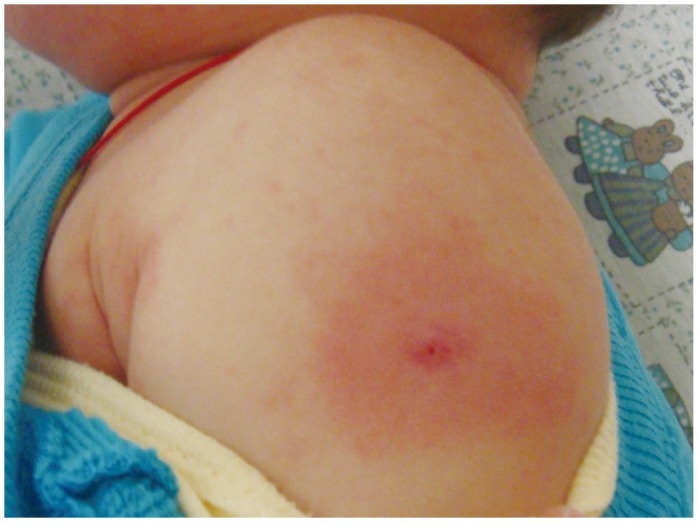
In addition to the diagnostic criteria, there is a broad range of nonspecific clinical features including severe irritability, insomnia, photophobia, fatigue, cough, vomiting, diarrhea, abdominal pain, anorexia, arthralgia, hypertension, tachycardia, and scarlet cheeks or nose. Complications include aseptic meningitis, uveitis, lymphadenitis, pneumonitis, gallbladder hydrops, urethritis, arthritis, hepatitis, nephritis, carditis, heart failure, and gangrene. Reactivation of a vaccine scar (Fig. 6), particularly with BCG immunization, is such a common clinical finding in East Asian countries where tuberculosis is endemic that it is included as a principle diagnostic feature. The principle features of KS may not always appear simultaneously, thus, making diagnosis difficult to piece together.
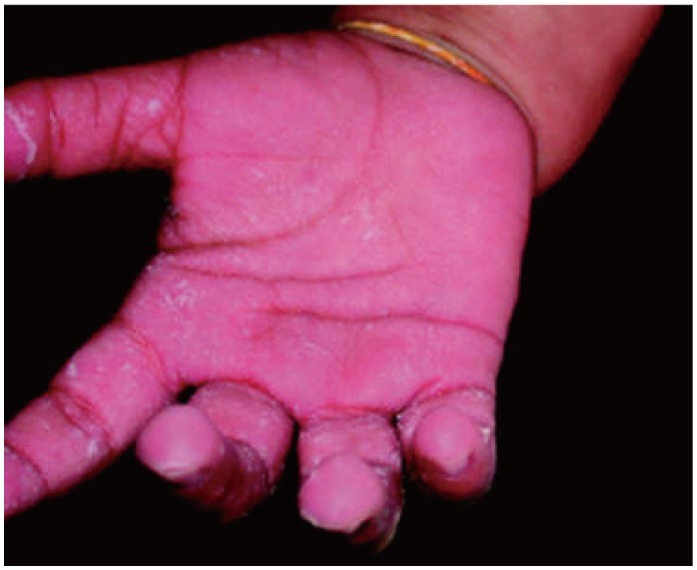
Unfortunately, there is no test to confirm KS as the cause of the illness remains unknown. KS continues to be diagnosed clinically and through the exclusion of other pediatric ailments, along with the aid of laboratory findings which are suggestive but not conclusive. Laboratory findings include a predominant leukocytosis, an elevated erythrocyte sedimentation rate, elevated C-reactive protein, elevated platelets, anemia, hypoalbuminemia, elevated alanine aminotransferase or aspartate aminotransferase, and sterile pyuria. KS may initially be mistaken for an infectious disease, particularly measles or scarlet fever. Infantile acrodynia is a childhood mercurial reaction (Figs. 7 and 8), which can mimic KS,1) and has led to its inclusion in the differential diagnosis of KS.2) Incomplete KS is diagnosed solely through the detection of coronary artery abnormalities when patients are noted with three or less principle diagnostic features. Patients with incomplete KS have the highest risk of coronary artery lesions (-95%) due to delayed diagnosis, and therefore it is very important to identify this subset of patients.
Kawasaki syndrome is regarded to be an autoimmune disease as opposed to an infectious one, although the cause is believed to be a widespread infection which is only pathogenic in susceptible children. Patients develop an autoimmune vasculitis in which the immune system attacks the blood vessels and induces systemic inflammation. Vasculitis shows a predilection for the coronary arteries which supplies oxygen-rich blood to the heart, and can result in the formation of coronary artery aneurysms and subsequent fatal thromboses. Therefore, it is of the utmost urgency that treatment with intravenous immunoglobulin (IVIG) be administered immediately after diagnosis of KS. Patients who do not respond to IVIG and remain febrile are typically treated with another dose of IVIG although some physicians will employ corticosteroids, tumor necrosis factor alpha (TNF-α) inhibitors (inflximab), or a combination of these treatments. In recent years, greater emphasis has been placed on the long-term follow-ups of patients as they are at risk for morbidity due to cardiac sequelae.
Over the last few decades in the developed world, KS has replaced rheumatic fever and become the leading cause of acquired heart disease. By far, the highest incidence of KS is found in Japan with an annual rate of 239.6 in 100000 (1 in 417.4) children under 5 years of age.3) At this current rate, more than 1 in every 100 Japanese children will develop KS before reaching adulthood. Following Japan, Korea and Taiwan have the next highest rates of KS with approximately 1 in every 200 and 1 in every 250 children respectively developing KS before adulthood.4)5) As prevalence of the illness continues to increase in Asian countries, it is also on the rise in many Western nations.
Genetics are believed to play an important role in KS because of several findings. Asian children, particularly of East Asian descent, are 10 to 20 times more likely to develop KS even if they reside in Western countries. Genetic clustering has been reported, while children with parents whom have experienced KS are twice as likely to develop the disease as compared to the general population. Siblings of children affected by KS are 10 times more likely to develop the illness than the general population, and this risk is even higher in twins regardless of whether they are monozygotic or dizygotic. Although KS is not contagious, more than 50% of these siblings develop KS within 10 days of the first sibling. As outlined by Onouchi,6) this can subsequently "... give the impression that KD is a type of endemic disease or a disease related to a lifestyle commonly found among the East Asian populations". While this no doubt reflects the shared genetic components conferring susceptibility, the role of exposure to specific environmental agents influenced by varying cultural habits, health practices, and economic or industrial development should not be overlooked. In this paper, we attempt to string these elements together to present an attractive gene-environment hypothesis for a mercury etiology in KS.
Inositol 1,4,5-Triphosphate 3-Kinase C
Over the last few years, the C allele of a functional single nucleotide polymorphism (SNP) of inositol 1,4,5-triphosphate 3-kinase C (ITPKC), rs28493229, was found to be associated with KS and subsequent coronary artery aneurysms in Japanese, Taiwanese, and US children.7-12) These findings were later supported by a genome-wide association study involving 2173 individuals with KS.7) One study of Taiwanese KS patients did not find any associations,13) although a later meta-analysis of Taiwanese studies confirmed a significant association of the ITPKC SNP in Taiwanese children with KS.8) An initial study of Han Chinese children had no associations to the ITPKC SNP,14) although in a subsequent investigation, the authors found that KS in Han Chinese patients was instead found to be associated with the ITPKC SNP rs2290692.15) A recent meta-analysis reviewing all the available patient data from previous studies reinforces a significant association between ITPKC and KS, and ITPKC currently remains the most consistently and strongly linked gene in KS (odds ratio=1.53).10)
Inositol 1,4,5-triphosphate 3-kinase C plays a role in the regulation of calcium ion signaling by converting inositol 1,4,5-triphosphate (IP3) to inositol 1,3,4,5-tetrakisphosphate (IP4). IP3 is a key second messenger molecule in various cell types which induces Ca2+ release from the sarcoplasmic reticulum by triggering IP3 receptors. ITPKC susceptibility in KS is believed to be a result of reduced genetic expression of ITPKC leading to inadequate phosphorylation of IP3, which promotes IP3 levels and subsequently increases Ca2+ release. In fact, the lowest genetic expression of ITPKC is observed in the Japanese followed by other East Asian ethnic populations. Knockdown and overexpression experiments indicate that ITPKC acts as a negative regulator of T and B cell activation through the Ca2+/NFAT pathway.6) As a result, a strong emphasis has been placed upon calcium signaling in KS and the Ca2+/NFAT pathway in particular. This recently led researchers to use calcineurin inhibitors in refractory patients with KS (n=10) who failed to respond to conventional, standardized treatments. Calcineurin inhibitors led to the rapid resolution of fever and inflammation in patients while appearing to be both safe and effective in the treatment of KS.16) However, ITPKC susceptibility passively confers susceptibility to KS and is believed to subsequently predispose patients to the etiological trigger of the disease.
This initially led us to consider the potential role of mercury and thimerosal in particular, an organomercurial preservative still used in certain pediatric immunizations which contains an oxidized mercury ion, as it is well known that Hg2+ is able to sensitize IP3 receptors.17-20) We recently discussed how ITPKC susceptibility in KS would make patients more susceptible to autoimmunity and coronary arterial wall relaxation from exposure to mercury or thimerosal.21) Ryanodine and IP3 receptors are vulnerable to the thiol redox state and are subsequently sensitized by low levels of Hg2+. Hg2+ induces a delayed, repetitive release of Ca2+ mediated by these sensitized receptors which has consequences for various cell types. Extracellular Ca2+ can also trigger these receptors, leading to calcium-induced calcium release. Empty intracellular Ca2+ stores are sensed by STIM1 which activates ORAI1 store-operated calcium channels, promoting further Ca2+ release. Mercury can also induce mitochondrial dysfunction, with mitochondria playing an important role in calcium regulation by storing intracellular Ca2+. Lastly, calcium influx by mercury would place additional stress upon the mitochondria which could also lead to dysfunction.
As mentioned earlier, the role of ITPKC in the regulation of calcium results in its role as a negative regulator of T and B cell activation. Ryanodine and IP3 receptors are expressed in various immune cells including T cells, neutrophils, macrophages, and dendritic cells which are all involved in the immunopathogenesis of KS. Certain immune cells are subequently vulnerable to sensitization by Hg2+ or thimerosal, in addition to subsequent increases in IP3 or Ca2+. Ca2+ also plays a key role in activating the platelets, with platelets often being elevated during KS. The resulting immune cell dysfunction can lead to autoimmunity, while immune cell infiltration of the endothelium could lead to systemic vascular inflammation and vasodilation. Previously, authors were unable to explain why ITPKC susceptibility would confer susceptibility to the development of coronary artery aneurysms in KS besides the implication that the activation of immune cells would lead to subsequent vascular inflammation. However, we recently discovered an aspect in which increased Ca2+ release would itself induce arterial vasodilation independent of the immune cell activation.21)
The ability of Hg2+ to induce endothelium-dependent vasodilation is attributed to its effects on prostaglandins, nitric oxide, and endothelium-derived hyperpolarizing factor (EDHF).18)22-26) Vascular wall relaxation by EDHF has been previously linked to the stimulation of calcium-activated potassium channels, which is another target of extracellular Ca2+ released from intracellular stores. Activation of these potassium channels leads to the hyperpolarization of smooth muscle cells, which results in their relaxation and the vascular wall expanding in diameter. The resulting hyperpolarization may then promote further relaxation by spreading electrotonically through the vascular wall via myoendothelial and homocellular smooth muscle gap junctions.18) In fact, thimerosal is found to fully relax contracted ring segments isolated from the coronary arteries.22) This could lead to the formation of coronary artery aneurysms observed in KS. Lastly, the neonatal myocardium is more sensitive to Ca2+ than that of adults, which would contribute to the predominant occurrences of KS in young children and the exceptional occurrence in adults.
Mercury increases levels of catecholamines such as norepinephrine and epinephrine which participate in the regulation of Ca2+ signaling. Therefore, we conducted a review of the effects that catecholamines would have on calcium influx mediated by ryanodine and IP3 receptors sensitized by Hg2+.
Catecholamines
Activation of catechol-O-methyltransferase (COMT) requires the donation of a methyl group from its coenzyme S-adenosylmethionine (SAM) and is dependent on its intact function. COMT degrades catecholamines such as dopamine, epinephrine, and norepinephrine by donating a methyl group to these catecholamines, resulting in their inactivation. Mercury binds to and inactivates SAM, resulting in the subsequent inactivation of COMT. Methionine synthase (MS) is also necessary for SAM formation, while mercury inhibits MS and decreases its activity, which results in reduced methylation. As a result of mercury disrupting to include the role of MS regulation on COMT accumulation of catecholamines such as norepinephrine and its metabolites (homovanillic acid, vanillylmandelic acid) occurs as catecholamines are not being properly degraded by COMT but instead persist for much longer.
Catecholamines are neurotransmitters and hormones which are centrally involved in the body's fight-or-flight response of the sympathetic system, and are primarily produced by chromaffin cells in the adrenal glands. An accumulation of catecholamines can lead to a clinical picture mimicking pheochromocytoma or neuroblastoma, as observed in infantile acrodynia.27-30) Catecholamines are frequently elevated in infantile acrodynia while many authors regard catecholamine levels to be more reliable in diagnosing acrodynia, as opposed to determining mercury levels in the blood or urine. Catecholamine-secreting tumors may also mimic the clinical picture of KS. Three children were reported of having a neuroblastoma during the acute phase of KS,31)32) which demonstrates that an accumulation of catecholamines may either exacerbate or trigger KS. Lastly, a SNP of the COMT gene (rs769224) was recently found to be significantly associated with the development of coronary artery lesions in KS.33)
β1-adrenergic receptors
Epinephrine and norepinephrine both bind to β1-adrenergic receptors with approximately the same affinity. Stimulation of β1-adrenergic receptors in cardiac cells leads to the phosphorylation of voltage-gated Ca2+ channels, which both opens the channels and extends the amount of time that these channels remain open. As a result, the permeability of the cardiac sarcolemma is increased, allowing Ca2+ to more readily enter the cell to trigger calcium-induced releases. At the same time, more Ca2+ released into the cytoplasm is able to exit the cell. Activation of β1-adrenergic receptors also leads to the phosphorylation of phospholamban, which enhances Ca2+-ATPase activity. Ca2+ is concentrated in the sarcoplasmic reticulum by Ca2+-ATPase, making more Ca2+ available for release. This causes a more forceful contraction, which increases arterial pressure and can lead to hypertension. Enhanced Ca2+-ATPase activity also rapidly removes Ca2+ from the cytosol by shortening the time Ca2+ is bound to troponin, causing shorter contractions which leads to tachycardia. A rapid heart rate would impair the ability of cardiovascular muscle to adequately relax as needed. Tachycardia or hypertension could lead to or promote the development of coronary artery aneurysms and subsequent thromboses, particularly in the tandem.
The kidneys also play a key role in the cardiovascular system, especially in adjusting blood pressure through its regulation of ions and water. Stimulation of β1-adrenergic receptors in the kidneys by norepinephrine or epinephrine increases renin secretion. Renin breaks down angiotensinogen to form angiotensin I, which is converted into angiotensin II by the angiotensin-converting enzyme (ACE). Angiotensin II is a potent vasoconstrictor affecting arteries and veins by raising blood pressure, while also stimulating the secretion of aldosterone and antidiuretic hormone (ADH) by the adrenal glands. Aldosterone and angiotensin II raise blood pressure by increasng water retention, Na+ reabsorption, and the secretion of K+. The formation of blood clots is promoted by angiotensin II through the adhesion and aggregation of platelets, while platelets are often elevated during KS. This could result in the formation of fatal thromboses as observed in KS or promote their development.
Polymorphisms of ACE have been associated with KS and subsequent coronary artery lesions.34-37) ACE levels are significantly lower during the acute phase of KS and normalize with convalescence, which is attributed to damaged endothelial cells during the course of the illness.38-40) Lower ACE levels would suggest that less angiotensin II is being formed, and subsequently less aldosterone and ADH would be formed. It is possible that lowering ACE levels could be a self-defensive physiological reaction in the body to reduce the formation of increased angiotensin II levels. Inhibition of ACE by captopril reduces serum matrix metalloproteinase-9 activity in KS patients and may prevent the subsequent formation of coronary artery lesions, particularly in patients who do not respond to IVIG.41) Therefore, it is likely that ACE susceptibility in KS is a result of increased genetic expression due to polymorphisms of the ACE gene, which in turn would lead to increased angiotensin II formation.
α1-adrenergic receptors
Both norepinephrine and epinephrine bind to α1-adrenergic receptors, although norepinephrine demonstrates a greater affinity. Stimulation of α1-adrenergic receptors activates phospholipase C resulting in the rapid formation of IP3 and diacylglycerol (DAG). Extracellular Ca2+ activates G-protein coupled receptors which increases catecholamine-induced formation of IP3. Myosin light chain is phosphorylated by IP3-released intracellular Ca2+ binding to calmodulin, and then activates myosin light chain kinase which enhances myosin ATPase activity. Through its production of protein kinase C (PKC), DAG also plays an important role in myosin light chain phosphorylation.42) Thus, contractions would occur more easily after Ca2+ binds to calmodulin, along with a subsequent relaxation after Ca2+ is released from calmodulin.
Stimulation of α1-adrenergic receptors by norepinephrine, extra- and intracellular Ca2+, calmodulin, and PKC also increases the secretion of atrial natriuretic peptide (ANP) by cardiac muscle cells, which is a potent vasodilator. ANP suppresses the release of vasoconstrictors such as renin, aldosterone, and ADH while also inhibiting the effects of catecholamines on smooth muscle tissues in arterioles and venules. Brain natriuretic peptide (BNP) and ANP increase renal excretion of water and Na+. This decreases blood volume and Na+ levels which reduces blood pressure. Upon autopsy, ANP expression was found to be moderately or remarkably increased in certain vascular lesions of patients with KS.43) BNP or N-terminal probrain natriuretic peptide (NT-proBNP) levels are frequently increased during the acute phase of KS and normalize during convalescence,44-53) and is associated with hyponatremia and the formation of coronary artery lesions.
Sodium
Hyponatremia is reported in 45% to 69% of KS patients53)54) and is associated with the formation of coronary artery lesions and giant coronary artery aneurysms.55)56) ADH levels are elevated in KS, which increases water retention and peripheral vascular resistance, hence, increasing blood pressure. At higher levels, ADH provokes hyponatremia due to the increased volume of water circulating in the blood. Syndrome of inappropriate antidiuretic hormone (SIADH) resulting from increased ADH levels are observed in KS and associated with hyponatremia.54)57)58) Levels of NT-proBNP were found to be higher in KS patients with hyponatremia, although NT-proBNP levels were lower in patients with SIADH,57) which is likely the result of BNP suppressing the release of vasoconstrictors including ADH. In KS, SIADH and increased ADH levels in KS could be a result of increased formation of angiotensin II by mercury which in turn would increase ADH and aldosterone levels. This may therefore also be a response to the diuretic effects of mercury, including via ANP and BNP. Mercurial drugs were widely used in medicine during the 19th century, particularly for their diuretic effects, while symptoms such as hypersalivation were unfortunately seen as proof of the efficacy of this toxic metal in the treatment of a myriad of ailments in children and adults. Lastly, it should be noted that hyponatremia is a frequent finding in infantile acrodynia and is believed to play a key role in the development of the illness and its subsequent symptoms.59)60)
Mercury can also inhibit Na+ uptake in skeletal and cardiac muscle cells through the occlusion of Na+ channels. As opposed to skeletal muscle cells which contain a single cysteine residue containing sulfhydryl with Hg2+ binding covalently to sulfur, Hg2+ prefers to bind covalently to the two cysteine residues in cardiac muscle cells to form a disulfide (S-Hg-S) bridge. This makes cardiac muscle far more sensitive to Hg2+ than skeletal muscle.61) The formation of these bridges block the entry and exit of Na+ through affected channels, which impairs or inhibits depolarization of these cells, which increases their possibility to hyperpolarize. This reduces or inhibits the ability for cardiac muscle cells to depolarize and generate potential to subsequently contract cardiac muscle or withdraw its relaxed state. In addition, oxidative stress by Hg2+ also impairs or inhibits contractions of smooth or cardiac muscle by compromising the ATP-dependent ability to bring Ca2+ back into the sarcoplasmic reticulum. Therefore, mercury has the ability to induce vasodilation of arteries through an influx of Ca2+ while encouraging the arteries to remain in a relaxed state. This could be intensified by mercury decreasing methionine synthase activity, which would reduce methylation and impair the body's ability to return to homeostasis, prolonging inflammation. This could be one way in which thimerosal is able to fully relax contracted ring segments of the coronary arteries as we mentioned earlier.
Genetic Synergism
We previously postulated that many genes are involved in KS and that they are likely to interact with one another by synergistically increasing the risk for KS and subsequent coronary artery lesions when presented together.21) In a recent study, KS patients expressing both ITPKC and caspase 3 (CASP3) polymorphisms were found to have a more significant risk of unresponsiveness to IVIG treatment than patients with only one of the SNPs.12) Polymorphisms of CASP3 have been associated with KS and subsequent coronary artery aneurysms.12)62)63) CASP3 promotes apoptosis, playing a key role in the apoptosis of immune cells, while a reduction in its expression would decrease immune cell apoptosis and prolong inflammation. In addition, CASP3 contains a cysteine residue making it vulnerable to covalent bonding with Hg2+.
One study discovered that the rs2229634 SNP of inositol 1,4,5-triphosphate receptor type-3 was associated with KS and subsequent coronary artery aneurysms.64) The inositol 1,4,5-triphosphate receptor type-1 (ITPR1) gene should also be investigated, as Hg2+ and thimerosal stimulate ITPR1. In addition, ITPR1 is expressed in immune cells such as dendritic cells which are involved in the immunopathogenesis of KS. As we mentioned earlier, Hg2+ and thimerosal sensitize both the ryanodine and IP3 receptors resulting in calcium influx. Genes related to ryanodine receptors could also be investigated, such as the RYR1 and RYR2 genes which are respectively expressed in skeletal and cardiac muscle. These genes should be investigated along with ITPKC to see if they are synergistic with one another.
Kuo et al.65) investigated polymorphisms of ORAI1, a gene that encodes calcium release-activated calcium channel protein 1 (CRACM1) which is essential in the function of store-operated calcium channels. ORAI1 also plays a key role in the activation of platelets and their subsequent aggregation at sites of vascular injury to form thromboses through store-operated entry of Ca2+ into platelets,66) while also having a crucial role in the function of T cell-mediated immunity and subsequent autoimmunity via store-operated entry of Ca2+ into T cells.67) In addition, ORAI1 is regulated via phosphorylation by PKC which suppresses store-operated Ca2+ channels and calcium-induced calcium release.68) The production of PKC is stimulated by norepinephrine and epinephrine, subsequently making ORAI1 an indirect target of Hg2+-induced increases in catecholamines. However, no association was observed between ORAI1 and KS risk while the authors suggested that a larger sample size should be investigated.65) It is likely that ORAI1 polymorphisms synergistically enhance the risks of KS and subsequent coronary artery lesions when expressed in combination with ITPKC. It is also possible that ORAI1 susceptibility may be largely or entirely dependent on being expressed in combination with ITPKC to even increase KS risk, as the gene is downstream from ITPKC.
In further support of the hypothesis that Ca2+ dysregulation plays a key role in the immunopathogenesis of KS, serum procalcitonin (PCT) levels were found to be increased in Japanese (n=25) and Spanish (n=9) patients with KS, while Japanese (n=2) and Spanish (n=1) patients who develop coronary artery aneurysms had PCT levels exceeding 3.0 ng/mL.69)70) Higher serum PCT levels were observed in Japanese patients with KS (n=160) who did not respond to IVIG compared to patients who responded to IVIG treatment.71) However, one study revealed no association between PCT levels and the risks of KS or subsequent coronary artery aneurysms in 18 French patients with KS,72) while another study indicated that low PCT levels (<0.5 ng/mL) in 38 Indian patients with KS were associated with the formation of coronary artery aneurysms.73) The calcitonin-related polypeptide alpha (CALCA/CALC1/CALC-I) gene encodes the peptide hormones PCT, calcitonin, calcitonin gene-related peptide and katacalcin which play a role in reducing Ca2+ levels. PCT is a precursor for calcitonin which reduces Ca2+ levels in the blood, while secretion of calcitonin is stimulated by increases in plasma Ca2+ levels. CALC1 susceptibility in KS would likely be the result of impaired formation of these peptide hormones by CALC1, and may synergistically increase KS risk when expressed in combination with ITPKC.
Of particular interest is the solute carrier family 8 (SLC8) gene, also known as the Na+/Ca2+ exchanger (NCX), which has three identified isoforms. SLC8A1/NCX1 is widely expressed in most tissues although it is most abundant in the heart, while expression of SLC8A2/NCX2 is restricted to the brain and spinal cord and SLC8A3/NCX3 expression is restricted to the brain and skeletal muscle.74) The Na+/Ca2+ exchanger is also expressed in immune cells, including SLC8A1/NCX1 and SLC8A3/NCX3 which are expressed in both macrophages and monocytes.75) NCX is a Na+-dependent Ca2+ antiporter which exchanges three sodium ions for a calcium ion. During relaxation in cardiac myocytes, Ca2+ is stored within the sarcoplasmic reticulum and mitochondria, although the overloading of these intracellular stores can result in oxidative stress and dysfunction. Therefore, cytosolic Ca2+ must be extruded from the cell, with NCX1 being the primary mechanism by which this is accomplished. This Na+/Ca2+ exchanger is the main manner in which cardiac myocytes are relaxed and returned to their resting state following excitation-contraction coupling, which increases intracellular Ca2+.76) Therefore, NCX susceptibility may potentiate autoimmunity and arterial wall relaxation resulting from calcium influx. Hyponatremia, which is often observed in KS patients, would likely impact this sodium-dependent calcium exchanger. Interestingly, placing macrophages expressing NCX1 or NCX3 in a Na+-free medium induced a marked release of TNF-α.75) During the acute phase of KS, TNF-α is elevated and plays an important role.
Discussion
As we have previously described, mercury has the ability to induce autoimmunity and arterial wall relaxation by sensitizing ryanodine and IP3 receptors, triggering delayed, repetitive calcium influxes which could lead to KS and subsequent coronary artery aneurysms. Mercury potentiating its own calcium influx through its promotion of catecholamine levels and subsequent IP3 formation may also have a synergistic effect, making it an even stronger candidate as a potential cause of KS. In addition, our review demonstrates that catecholamines may play an important role in the development and severity of autoimmunity and subsequent coronary artery aneurysms in KS. Unfortunately, it appears that catecholamines have not been investigated in KS patients or in the pathogenesis of KS.
Thimerosal, an organomercurial preservative used in vaccines and other medical biologicals, would have a similar effect as it degrades into thiosalicylate and ethylmercury, the latter of which contains Hg2+. It is reasonably safe to assume that methylmercury, which also contains Hg2+, would have a similar effect, although this requires further investigation. In the brain, disruption of Ca2+ signaling is an important mechanism in which methylmercury is able to induce neurotoxicity, and one manner in which methylmercury accomplishes this is through the sensitization or inhibition of IP3 receptors.76)77) The global problem of methylmercury in the contamination of fish may help explain why KS is overexpressed in children of Asian descent, as they are to 10 to 20 times more likely to develop KS, even when they reside in Western nations.
Our review provides several areas of investigation for KS regarding the immunopathogenesis of the illness and the role of genetics. Genes involving catecholamines or calcium signaling could be investigated, particularly to see if there are any synergistic effects with ITPKC and other genes. The serum and urine of patients could be investigated to see if catecholamines such as norepinephrine, epinephrine, dopamine, and their precursors or metabolites are elevated. Levels of vasoconstrictors, vasodilators, and their precursors which are also involved in Na+ regulation could be investigated, including the renin-angiotensin system, aldosterone, ADH, ANP, and BNP.
Mercury levels in the urine, blood, or hair from KS patients could also be investigated, although they are unreliable in the diagnosis of acrodynia due to mercury's rather mercurial behavior. Previous investigations have found elevated mercury levels in patients with KS78)79) while others found normal levels.81) Mercury excretion by an individual varies greatly, which is why a 24-hour urine collection is preferred, whereas day-to-day levels are still highly erratic and can differ immensely. More importantly, sources of mercury exposure should be investigated and compared to mercury levels in the urine, blood, and/or hair.
Although now declining in use, dental amalgam fillings continue to be the predominant source of total mercury body burden in humans (70-95%) and are the largest source of maternal mercury with regards to the in utero exposure of the unborn child.82) Immunizations may shortly precede the onset of the KS although no associations have been established. This highlights the necessity to study the risks of KS and subsequent coronary artery lesions between vaccinated and unvaccinated children, while the role of the vaccine additives such as thimerosal and aluminum should also be examined. An investigation comparing the rates of KS and exposure to methylmercury from fish consumptions among various ethnic groups with genetic and cultural influences in mind could be very interesting.
Conclusion
We conclude that there is biological evidence which demonstrates that mercury is able to trigger a pathogenic process which results in autoimmunity and coronary artery lesions as observed in KS, while the ITPKC polymorphism in KS would also make patients more susceptible to mercury via its impact upon calcium signaling. Further investigation of mercurial etiology of KS is justified, since no infectious cause has been established over the past four decades. The confirmation of mercury as a cause of KS could lead to the discovery of additional etiological triggers such as infections, drugs, or other environmental toxins. Other xenobiotics such as infections may contribute to oxidative stress and synergistically enhance the effects of mercury, particularly xenobiotics which impact calcium signaling.
Influencing calcium signaling is one major way in which allergens induce a hypersensitivity response, while mercury and thimerosal are recognized allergens which increase IgE levels. Genotypes conferring risk to KS in addition to idiosyncratic and allergic hypersensitivity reactions may explain why most children appear to be able to tolerate mercury, while others develop a mercurial reaction induced by a subtoxic dose of the metal. This would agree with the observations of the acrodynia epidemic during the 1800s and ending in the 1950s, during which mercury exposure was common with the use of calomel and teething powders, although only 1 in every 500 children had developed the disease. However, calomel (mercurous chloride) is an inorganic mercurial which is 100-fold less toxic in vitro than the organomercurial preservative, thimerosal.83)
Methylmercury exposure from fish consumption may contribute to overexpression of the illness in Asian children who are far more likely to develop KS, even if they reside in Western countries. Increasing methylmercury contamination in fish is a byproduct of an alarming rise in global anthropogenic mercury pollution, with a dramatic 3- to 5-fold increase over the last few decades alone.84) This demonstrates the urgent need to further reduce mercury emissions, while increasing global rates of mercury pollution parallels the increasing incidence of KS in Japan when epidemic years (1979, 1982, 1986) were excluded. Avoidance of tuna fish and other predatory fish is highly recommended for young children and pregnant women. This lack of an important source of protein and essential fatty acids should be supplemented, including by incorporating a diet of fish and seafood containing low levels of methylmercury.
Mercury is found to be toxic in every chemical form and is 10 times more toxic than lead (Pb) in vitro.85)86) A safety level cannot be established for mercury as susceptible individuals react to doses which are considered to be subtoxic. Mercury has no physiological role in the human body, and we are reminded by Martindale87) that mercury "... should not be given to infants or applied to their skin as they may cause acrodynia". Eli Lilly, the manufacturer and patent owner of thimerosal, specifies in their safety information that the mercurial "... may cause severe mental and motor retardation in children and the unborn". The entirely unnecessary, ongoing iatrogenic use of mercurials in medicine should be fully discontinued, particularly within the use of thimerosal in pediatric vaccines. However, the ongoing use of dental amalgam tooth fillings, which continuously emits mercury vapor, remain the largest source of mercury in the human body (70-95%) while also contributing significantly to environmental mercury pollution.82)
 XML Download
XML Download