

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Breast cancer is the most common type of cancer affecting women (in 154 countries worldwide) and the most frequent cause of cancer mortality in women (in 103 countries worldwide) [1]. More than 2 million women were diagnosed with breast cancer in 2018, and more than half a million women succumbed to the disease [1].
Approximately 10% of the breast cancer cases occur in women who have a first-degree relative with a history of breast cancer [2]. However, underlying inherited gene mutations, that could explain for familial clusters, could only be identified in 20%–25% of these families (genes, that were tested: BRCA1, BRCA2, TP53, PTEN, ATM, CHEK2, and HRAS1) [34]. Mutations in BRCA1 and BRCA2 constitute the largest proportion of these [3]. The life time risk of breast cancers in carriers of a BRCA1 mutation is up to 70% compared to around 12% in non-carriers [4].
Tumors of the breast epithelium can be further divided into different subtypes based on their gene expression profiles. The basal-like subtype of breast cancer (basal-like breast cancer [BLBC]) accounts for up to 75% of all breast cancers in BRCA1 mutation carriers and is generally associated with an inferior prognosis of survival [56].
Given that BRCA1's first cloning in 1994 followed genetic analysis of patients from familial clusters, it was expected that its cellular function would explain its tissue-specificity. Surprisingly, the first insights into BRCA1 biochemistry revealed its involvement in DNA double strand break (DSB) repair mechanisms which are required in all tissues. Ongoing research has unveiled a network of BRCA1's interactions in the cell and several concepts accounting for the tissue-specificity have emerged. Here, we have summarized these in the light of recent advances in breast tumorigenesis.
Firstly, a short overview of BRCA1's cellular interactions is provided. Then a model of tissue-specificity of cancer genes by Schaefer and Serrano [7] is presented and applied to BRCA1 to review the proposed concepts. Finally, we outline the impact of these concepts on the formation of hormone-sensitive and -insensitive types of breast cancers, discuss how they might account for other typical features of BRCA1-associated breast cancer, and present possible pharmacological interventions.
BRCA1 PROTEIN STRUCTURE AND CELLULAR INTERACTION
The cellular function of BRCA1 is heavily reliant on its interaction with other proteins (Figure 1). In view of the complexity stemming from the involvement of BRCA1 in different protein complexes, this article will only provide a short overview of interactions focusing on aspects that are relevant in the following passages. Several other reviews can be considered for more detailed insights [891011].
Figure 1
Domains of and selected proteins that interact with BRCA1.
BARD1 = BRCA1-associated RING domain protein 1; PALB2 = partner and localizer of BRCA2; CTIP = C-terminal binding protein interacting protein; ABRAXAS = BRCA1 A complex subunit; BACH1 = BTB domain and CNC homolog 1; BRCT = BRCA1 C-terminus.
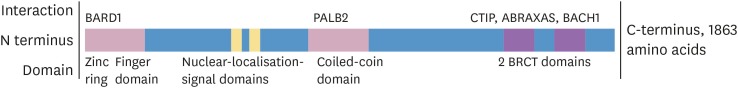
BRCA1's involvement in the maintenance of genomic integrity mainly relies on its central role in protein complexes that are required for the repair of DSB and stalled replication forks [8]. It is a central component of several protein complexes that are required for these tasks. Via its BRCA1 C-terminus domains—highly conserved motifs expressed in several proteins involved in DSB repair—BRCA1 facilitates assembly of complexes with proteins including BTB domain and CNC homolog 1, retinoblastoma binding protein 8 and BRCA1 A complex subunit that have distinct functions in the recognition and initiation of DSB repair [11]. The BRCA1-BRCA1-associated RING domain protein 1 (BARD1) complex acts as an E3-ubiquitin ligase, an enzyme capable of transferring ubiquitin molecules from E2 ligases onto specific substrates [9]. BRCA1-BARD1 complex-mediated ubiquitination of histone H2A has been suggested to be necessary for the repositioning of tumor-suppressor p53-binding protein 1 from the sites of DSB [12]. This is thought to favor homologous recombination (HR)-mediated repair of DSBs instead of the error prone alternative non-homologous end joining [1112]. The BRCA1-partner and localizer of BRCA2-BRCA2 complex is necessary for the recruitment of RAD51 recombinase (RAD51) to the site of DSBs [8]. RAD51 itself forms a protein complex that mediates the search for homologous DNA sequences and consequent strand invasion initiating the repair process [8911].
In addition to DSB repair, BRCA1 serves an important role in the regulation of transcription. Since BRCA1 lacks any specific DNA binding domains, this function is mediated indirectly via either the modulation of transcription regulating pathways or direct protein-protein-interaction with transcription factors [10]. For example, BRCA1 directly interacts with the estrogen receptor ([ER]; although this mostly refers to the α-subtype, participation of ERβ cannot be ruled out because some references have not provided distinctions between the different subtypes) inhibiting its transcriptional activity [10].
A UNIVERSAL MODEL OF TISSUE-SPECIFICITY OF CANCER GENES
In 2016 Schaefer and Serrano [7] analyzed the association between mutations in certain genes and their tissue-specificity. They described 4 categories of possible mechanisms: 1) tissue-specific differences in the expression of the gene that result in predisposition of certain tissues; 2) expression of a different, functionally redundant protein, that compensates for the lack in unaffected tissues; 3) cell-type specific functions of the gene that lead to the tissue-specificity, and 4) tissue-specific cell properties or environmental factors that could drive local tumorigenesis. In this review, we have employed this model to provide a systematic overview of the findings from ongoing research on BRCA1 to aid in our understanding of the mechanisms underlying its tissue-specific tumorigenicity.
TISSUE-SPECIFIC EXPRESSION OF BRCA1
Cell type-specific differences in BRCA1 protein expression levels (“expression” and “expression level” will subsequently refer to protein expression levels if not accompanied by a note) could account for specific behavior of cells harboring 1 mutant BRCA1 allele (subsequently termed haplodeficient cells).
Modern advances in fully automated cell expression analysis have facilitated the development of detailed databases for human tissue-specific mRNA and protein levels. Evaluation of BRCA1 mRNA and protein expression using the Human Protein Atlas database (available under https://www.proteinatlas.org/) reveals no difference in the levels of either BRCA1 mRNA or protein in breast epithelial cells (BECs) compared to that seen in tissues not affected by BRCA1 haplodeficiency. For example, the measured transcripts per million of BRCA1 mRNA were 4.4 in BECs and around 10 in tissues of the immune system such as lymph nodes or tonsils (data by https://www.proteinatlas.org/ENSG00000012048-BRCA1/tissue, accessed August 2018) [13].
As expression levels are insufficient to explain tissue-specificity, alternative splicing becomes a subject of interest. Today, more than 50 different mRNAs derived through alternative splicing from the BRCA1 gene locus have been identified in humans of which 10 exhibit relevant expression levels (> 5% of the expression levels of the full-length transcript) [1415]. Initial evaluation of splice variants in human BECs and human whole blood samples did not reveal any difference in the levels of expression [14]. Additional data comparing levels of expression in breast epithelium with other tissues was not found.
In summary, total protein and mRNA expression levels of BRCA1 do not provide an explanation for its tissue-specificity. The role of alternative splicing is not conclusive yet, but initial evidence indicates no tissue-specific expression of splice variants in human BECs.
TISSUE-SPECIFIC EXPRESSION OF REDUNDANT PROTEINS
Tissue-specific expression of proteins, that are capable of compensating for BRCA1 haplodeficiency in non-breast tissues, but lack of expression of these proteins in human BECs, could also account for the predisposition of breast tissue to BRCA1-associated tumorigenesis.
Unfortunately, there is little research addressing this possibility and due to BRCA1's interaction in many different pathways, several lines of research will be required to evaluate tissue-specific redundancy for every distinct function.
Nonetheless, it has recently been shown that human skin keratinocytes and fibroblasts use DNA repair pathways differently in response to DNA damage [16]. The cells were found to use different subtypes of nucleotide excision repair to repair ultraviolet induced DNA lesions: fibroblasts solely relied on the transcription coupled repair subtype whereas keratinocytes additionally exhibited the global genome repair subtype [16].
Likewise, cell-type specific differences in the usage of DSB repair pathways and in other BRCA1-specific interactions might exist although there is no supporting evidence to date.
TISSUE-SPECIFIC FUNCTION OF THE CANCER GENE
Regulation of the activity of aromatase
Synthesis of estrogen is dependent on the key enzyme aromatase. In addition to the granulosa cells in the ovaries, which synthesize a major fraction of the systemic estrogen, other tissues contribute to plasma levels via aromatase-mediated synthesis [17]. In vitro experiments have revealed that the expression of aromatase is negatively regulated by BRCA1 [1819] (Figure 2 upper left). Consistently, plasma estrogen concentrations in BRCA1 mutation carriers have been reported to be elevated by as much as 30% [20]. Interestingly, the same study has reported peak progesterone levels to be elevated by as much as 121% in these carriers, although no interaction of BRCA1 and progesterone synthesis was identified.
Figure 2
The interaction of different concepts for tissue-specific tumorigenesis in BRCA1 mutation carriers.
All arrows behind proteins or processes indicate their change of activity in comparison to non-mutated individuals (aromatase ↑ indicates that its activity in mutation carriers is increased). Arrows between proteins or processes indicate whether the interaction increases or decreases the activity of the affected participant (BRCA1 ⟞ aromatase indicates that BRCA1 decreases aromatase's activity; consequently BRCA ↓ ⟞ aromatase ↑ indicates that 1) BRCA1 is decreased in mutation carriers, 2) that it physiologically decreases aromatases activity, and 3) that aromatase's activity is subsequently increased in mutation carriers). Not all concepts are illustrated. For detailed information see text.
ES = estrogen; ER = estrogen receptor; PO = progesterone; PR = progesterone receptor; NF-κB = nuclear factor-κB; NRF2 = nuclear factor (erythroid-derived 2)-like 2; RANKL = receptor activator of nuclear factor-κB ligand; JAG1 = jagged 1; CYP1A1 = cytochrome P450 1A1; SIRT1 = sirtuin 1; SNAI2 = snail family transcriptional repressor 2.
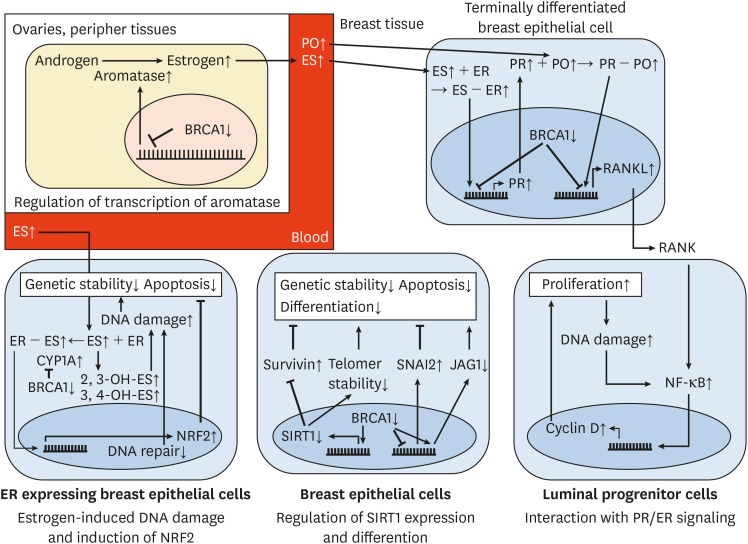
A correlation between estrogen levels and breast cancer has been noted in BRCA1 non-mutated individuals [21]. Several concepts that will be discussed in this article, in addition, suggest a participation of estrogen and progesterone in tumorigenesis. However, the efficiency of estrogen lowering therapy—oophorectomy or pharmacological estrogen antagonism—in BRCA1 mutation carriers is a topic of intense discussion [22232425].
Estrogen and progesterone signaling
The epidemiologic observation that tumorigenesis in BRCA1 mutation carriers is almost exclusively limited to the ovaries and breast tissue, both of which are under the influence of sex hormones, has led to intensive research on possible interactions between BRCA1 and sex hormone signaling [26]. This soon led to the discovery of mutual regulation highlighting a possible involvement in tissue-specific tumorigenesis. Specifically, the signaling mediated by ERs and progesterone receptors (PRs) is repressed by BRCA1 through transcriptional repression of the receptors as well as direct protein-protein interaction [10]. Additionally, ER signaling enhances BRCA1's transcription and increases the expression of PR [27].
This brought up a model in which BRCA1 deficiency leads to increased activity of ER and PR, sensitizing cells for sex hormone-dependent proliferation thereby promoting tumorigenesis. However, up to 75% of breast tumors arising in BRCA1 mutation carriers are BLBCs and more that 80% do not express ER or PR [56]. New approaches were therefore devised to unravel the underlying mechanisms.
Firstly, experiments in a mouse model suggested that initiation of tumorigenesis takes place in ER sensitive cells which then lose their ER expression [28]. This could occur through positive selection of cells that acquire estrogen-independent proliferation through further mutations. Loss of the ER provides these cells with further advantages as ER plays a role in the induction of DNA repair genes [282930]. Such a loss of a tumor initiating gene has already been observed in different settings [3132]. Additionally, BRCA1's role in differentiation (as described in the following section ‘Regulation of BEC differentiation’) provides insights into possible mechanisms that might lead to the loss of ER expression.
Recent research on the hierarchy of BEC differentiation has, however, suggested that a specific subpopulation of luminal progenitor cells (LPCs)—defined by the expression of receptor activator of nuclear factor-κB (RANK) but lacking expression of both ER and PR—is especially prone to be the cell of origin for BLBCs [3334].
It was, therefore, proposed that the influence of ER and PR signaling on tumorigenesis is mediated through paracrine mechanisms [2730] (Figure 2, right side, terminally differentiated cell and LPC). Terminally differentiated BECs, that express ER and PR, exhibit paracrine influences on LPCs thereby positively regulating their proliferation [27]. This is mediated by PR-induced transcription of RANK ligand (RANKL) which is highly upregulated in BRCA1 haplodeficient BECs (an increase greater than 5-fold was reported) [35].
RANKL itself binds to its receptor RANK that in the human breast tissue is uniquely expressed on the subpopulation of LPCs mentioned above [33]. This results in activation of the transcription factor nuclear factor-κB (NF-κB), that enhances transcription of genes favoring proliferation (e.g. cyclin D1) [27].
The proliferation associated DNA damage in LPCs and insufficient repair of these damages in BRCA1 haplodeficient cells were shown to further increase NF-κB activity resulting in even greater proliferation of these cells [27].
Histological examination of breast tissue from BRCA1 mutation carriers confirmed that the number of LPCs was increased by as much as a factor of 2 in comparison to that in non-mutated individuals [2736]. Furthermore, these LPCs displayed greater vulnerability to DNA damaging agents such as hydroxyurea than LPCs from healthy controls [33]. Interestingly, even compared to other BECs from mutation carriers, RANK expressing LPCs display increased vulnerability to DNA damage [33].
Regulation of breast epithelial cell differentiation
NOTCH and snail family transcriptional repressor 2 (SNAI2, commonly known as SLUG) are regulators of differentiation in the breast tissue that show important interactions with BRCA1 (Figure 2, lower middle, BEC) [3738].
BRCA1 interacts with NOTCH signaling via transcriptional upregulation of jagged 1 (JAG1)—a ligand of the NOTCH receptor [37]. Binding of JAG1 to NOTCH results in a proteolytic cleavage of the NOTCH intracellular domain which then alters transcription favoring differentiation.
Expression of SNAI2 on the other hand is negatively regulated by BRCA1 [38]. Since SNAI2 mediates stem cell like behavior, BRCA1-dependent repression of SNAI2 also favors differentiation.
Accordingly, in vitro siRNA-mediated knockout of BRCA1 almost completely freezes the differentiation of breast progenitor cells such as LPCs as well as earlier progenitor cells termed basal stem cells [3739]. It leads to diminished expression of luminal markers, most markedly ER expression, and enhanced expression of proliferation-associated markers (e.g. Forkhead Box M1 or C-X-C motif chemokine ligand 1) as well as markers typically expressed on basal stem cells (e.g. p-cadherin, also known as Cadherin 3 or CTP synthase 1) [37]. Nonetheless other typical progenitor cell markers as Nanog homeobox are decreased in these cells implying that BRCA1 deficiency does not lead to physiological dedifferentiation but rather causes the formation of aberrant progenitor cells [37].
The level of differentiation was proposed to depend on the levels of BRCA1 and haplodeficiency was suggested to arrest differentiation at the level of LPCs [40]. However, this has not yet been tested experimentally.
In addition to this increase in dedifferentiated progenitor cells that might undergo tumorigenesis, BRCA1's role in the expression of ER is important. As mentioned above, ER might be expressed during tumor initiation and then lost while further progression [28]. Decrease of BRCA1 expression in genetically unstable early malignant cells, subsequent dedifferentiation and loss of luminal markers (including ER) might explain this observation [30].
CELL-TYPE SPECIFIC PROPERTIES AND TISSUE ENVIRONMENT
Tissue-specific rates of loss of heterozygosity
The human genome is composed of pairs of homologous chromosomes. These copies are mostly identical with a difference in about 0.6% of the base pairs. The spots with differences in the DNA sequence are called heterozygote spots and are widely spread among the DNA.
Loss of heterozygosity (LOH) in a specific area has been among the first observations of genomic changes in cancer. LOH could lead to the loss of the second allele of a tumor suppressor in hereditary haplodeficiency which according to Knudsen's second hit hypothesis is the initiating step in tumorigenesis.
This second allele can be lost by either translocation/inversion (copy-neutral LOH) or loss of genetic material (parts of chromosomes, whole chromosomes; LOH with change of copy number). Another possible mechanism for LOH is DSB repair by HR. This requires a template to repair the DSB and can use either the homologous chromosome or the sister chromatid (only available after replication of the cell) [41]. When using the homologous chromosome (interchromosomal HR), this changes the original sequence in a process similar to crossing-over [41].
Usage of interchromosomal HR to repair DSB was suggested to provide insights into tissue-specific occurrence of LOH [42]. Tissue-specific higher expression of BRCA1 in breast tissue was proposed to require a more relaxed chromatin environment which favors use of interchromosomal HR for repair of DSBs in this region [42]. This would then cause increased occurrence of LOH at this locus and therefore promote tumorigenesis in a tissue-specific manner [42].
However, this hypothesis has never been tested experimentally, interchromosomal HR has been proven to be a rare event in human somatic cells [41] and expression of BRCA1 is not tissue-specific (see section about BRCA1 expression). Nonetheless, the rates of LOH have been found to be variable in different murine cell types and mechanisms other than interchromosomal HR might contribute to similar differences in rates of LOH in humans [43].
Interaction with X inactivation specific transcript RNA
X inactivation specific transcript (XIST) is a noncoding form of RNA that is required for the inactivation of the second X chromosome in cells of females to maintain correct gene dosage [44].
BRCA1 has been implicated for proper formation of XIST RNA [454647]. Insufficient coverage of the second X chromosome in BRCA1 deficient carriers was proposed to be a cause of tumorigenesis [454647]. According to this model, tissue-specificity arises via increased transcription of X-chromosomal genes, that are involved in breast tumorigenesis. This is backed by observations suggesting that X gene dosage is important in the development of breast cancer [48].
Three independent publications in 2007 have, however, shed doubt on the proposed involvement of BRCA1 in XIST-mediated coverage of the X chromosome [495051]. Neither in vitro experiments [4951] nor examination of breast tumor samples of BRCA1 haplodeficient individuals [50] confirmed a correlation between BRCA1 gene status and sufficient XIST coverage. Instead of the originally proposed direct interaction, the influence of BRCA1 on the coverage of the second X chromosome was suggested to arise from its broad role in maintaining genomic integrity [45].
Estrogen-dependent induction of NF (erythroid-derived 2)-like 2
Human cells undergoing tumorigenic transformation experience high levels of oxidative stress that lead to DNA damage and increased likelihood of apoptosis thereby limiting carcinogenic growth [52].
One important component of the cellular oxidative stress response is the NF (erythroid-derived 2)-like 2 (NRF2). NRF2 controls expression of proteins relevant for induction of antioxidative proteins and hence counteracts oxidative stress [53].
BRCA1 enhances the activity of NRF2 by stabilizing and activating it [54] (Figure 2, lower left, ER expressing BEC). BRCA1 deficient cells therefore express reduced levels of NRF2 [54]. Whether this is also applicable to BRCA1 haplodeficient cells, has not been tested yet. Consequently, BRCA1 deficient cells, that undergo tumorigenic transformation and associated oxidative stress [53], exhibit earlier apoptosis.
However, estrogen also elevates NRF2 levels. This effect is mediated via its binding to the intracellular ER, subsequent activation of the phosphatidylinositol-3-OH kinase [55], and activation of AKT signaling resulting in enhanced translation of NRF2 [53]. In the breast tissue, high concentrations of estrogen might thereby enhance survival of cells undergoing tumorigenesis and promote the progression into hormone-sensitive tumors. BLBCs, that arise in ER-negative LPCs, however are not likely to be affected.
Estrogen-induced DNA damage
Estrogen has multiple effects on the breast tissue including an increase in DNA damage resulting from an elevated level of DNA damaging molecules generated by several mechanisms (Figure 2, lower left, ER expressing BEC).
Estrogen-inactivating pathways release genotoxic metabolites [56]. These include 2-hydroxyestradiol (2-OH-E2) and 4-hydroxyestradiol (4-OH-E2) which are generated from estradiol (the most potent of the different estrogens) by CYP450-class enzyme dependent oxidation (e.g. cytochrome P450 1A1 [CYP1A1]). The 2-OH-E2 and 4-OH-E2 are then further metabolized into quinones (by the same enzymes) which react with DNA molecules to form covalent adducts with purine bases [56]. In BECs the flow through this pathway is especially high because of the elevated estrogen levels in the tissue that exceed plasma levels [5758]. Additionally, BRCA1 regulates expression of the estrogen metabolizing enzymes (e.g. CYP1A1) in a tissue-specific manner [56]. Whereas BRCA1 haplodeficient BECs express higher levels of CYP1A1 (in comparison with nondeficient BECs), enzyme levels in kidney cells are not affected by BRCA1 deletion [56]. Increased CYP1A1 levels accelerate 2-OH-E2 and 4OH-E2 formation and subsequent DNA damage.
Many of the BRCA1-mediated cellular functions (e.g. DSB repair or cell-cycle checkpoint control) have been found to occur at similar levels in BRCA1 haplodeficient cells compared to non-mutated cells [59]. However, BRCA1 haplodeficient cells are more vulnerable to replicational stress [59]. In the breast tissue, estrogen-induced high concentrations of genotoxic metabolites are thought to create increased replicational stress leading to genetic instability [5660].
Tissue-specific regulation of sirtuin 1
Sirtuin 1 (SIRT1) is an enzyme capable of deacetylating a huge variety of cellular proteins. It is therefore involved in a lot of different cellular functions (e.g. energy metabolism, regulation of differentiation, and DNA damage repair) [616263].
Especially important for the discussion in this article is its function in the regulation of telomeric length and in the induction of apoptosis. Firstly, SIRT1 is an inhibitor of the anti-apoptotic protein survivin [64]. Secondly, SIRT1 expression is necessary for elongation of telomers and decreased cellular expression results in telomer shortening [6365]. Although the exact mechanism of this interaction still needs to be elucidated, evidence suggests a positive interaction with telomerase activity, increased stability of telomeres by deacetylation of certain histones (histone 4 lysine 16 deacetylation seems to be highly significant in this context) and enhanced activity of proteins involved in HR (e.g. Nibrin, Werner syndrome RecQ like helicase) [63]. As decreased telomer length correlates with genetic instability, SIRT1 performs an important role in the maintenance of genomic integrity [66].
The transcription of SIRT1 is positively regulated by BRCA1 in a tissue-specific manner [67] (Figure 2, lower middle, BEC). Examination of SIRT1 expression in BRCA1 haplodeficient human BECs, human dermal fibroblasts, human mammary fibroblasts and human primary keratinocytes has revealed diminished expression in the BECs. Levels in the other cell types were, however, not found to differ in comparison to BRCA1 non-mutated cells [67].
CONCLUSION
The seemingly paradox notion that despite its role in DSB repair, BRCA1 mutation carriers almost exclusively develop breast cancer at a higher frequency, is being gradually solved with research findings that are helping us understand the broad integration of BRCA1 with cellular functions including differentiation, proliferation and metabolism. Whereas some concepts—tissue-specific level of BRCA1 expression or redundant proteins, interaction with XIST RNA, tissue-specific rates of LOH—could not be confirmed, others have provided insights into underlying mechanisms.
Although hormone-sensitive breast tumors and BLBCs both arise from BECs, different mechanisms seem to promote their occurrence. Hormone-sensitive tumorigenesis, occurring in ER and PR positive cells [34], is accelerated by estrogen-induced NRF2 expression and estrogen dependent formation of genotoxic metabolites. BLBCs arising in ER negative LPCs on the other hand are promoted by RANKL dependent expansion and increased proliferation of the LPC population. Dedifferentiation and stimulation of stem-cell like behavior as well as downregulation of SIRT1 contribute to tumorigenesis in both populations.
Interestingly, the concepts mentioned above could also help to understand 2 other observations. Recent investigations question whether LOH at the BRCA1 locus is necessary to initiate tumorigenesis, which counters the widespread Knudson hypothesis [6869]. Indeed, several of the approaches discussed above (e.g. SIRT1 expression, repair of DNA damage, differentiation or RANKL expression) do not rely on the loss of both alleles of BRCA1. Instead, haplodeficiency for BRCA1 is sufficient to severely restrict physiological cellular behavior. Additionally, the notion that somatic BRCA1 mutations or epigenetic silencing is less frequent in sporadic breast cancer (between 10% and 20% of all cases; up to 50% showed genetic alterations of p53) counters its outstanding association with hereditary forms. Considering the complex interaction of multiple cell-types in BRCA1 associated breast tumorigenesis, which somatic mutations cannot provide, helps with the understanding of this observation.
Further investigation will hopefully elucidate the underlying mechanisms in detail and might ultimately help to advance medical interventions for mutation carriers. As in vitro studies confirmed that antibodies against RANKL abrogated the ability of BRCA1 haplodeficient cells to survive and proliferate, denosumab is currently undergoing clinical evaluation for the prevention of breast tumors in BRCA1 mutation carriers [3335].
In this review, we have summarized the mechanisms underlying the predisposition for breast cancer in BRCA1 mutation carriers. The tissue-specificity has nothing to do with the role of BRCA1 in DSB repair. Moreover, neither the tissue-specific expression of BRCA1 nor the absence of redundant proteins could provide insights into this observation. Instead, it arises from the synergy of BRCA1's interactions unique to BECs and a promoting environment shape in the breast tissue.
 XML Download
XML Download