

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Congenital myopathies (CMs) are a heterogeneous group of inherited muscle disorders that are divided into subtypes based on the predominant histopathologic findings, namely nemaline rods, cores, central nuclei, or fiber-type disproportion.12 However, the clinical and pathologic phenotypes are not always correlated with the genotype in CM. The same genetic mutation can give rise to different phenotypes, and more than two genes can cause the same histopathologic findings.3 Additionally, the application of next-generation sequencing improves the molecular diagnosis of genetic disorders, including CM. Mutations in more than 45 causative genes have so far been implicated in CM,4 and recent studies have frequently defined subtypes based on causative genes and analyzed the clinical and pathologic genetic subtypes.156
RYR1-related myopathy is one of the most common CMs.5678
RYR1 encodes ryanodine receptor 1 (RYR1), which is the principal channel for releasing calcium in the sarcoplasmic reticulum plays a crucial role in excitation-contraction coupling. The classical clinical features of RYR1-related myopathy have been recognized as central core disease (CCD) and malignant hyperthermia (MH) susceptibility. However, recently reported RYR1-related myopathies include forms of not only CCD but also multiminicore disease, centronuclear myopathy, type-1 predominance, and congenital fiber-type disproportion (CFTD).9 We evaluated the characteristics of RYR1-related myopathy in a Korean population by analyzing clinical, pathologic, and genetic findings obtained from five unrelated patients with RYR1 mutations.
METHODS
Subjects
We reviewed the medical records of a myopathy database from January 2002 to December 2014. Five unrelated patients with RYR1 mutations (ID25, ID37, ID45, ID131, and ID185) were identified and included in this study. We have reported on these patients previously, but did not demonstrate detailed clinical and pathologic phenotypes.10 The present research protocol was approved by the Institutional Review Board of Gangnam Severance Hospital, Korea (IRB number: 3-2017-0051). All participants had previously been required to sign an informed-consent document for the use of their data genetic analysis.
Variant analysis
All variants were classified according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines.11 We filtered polymorphisms found in the Korean population (n=298) while also using public databases [dbSNP 135 and 1000 Genome project SNP (2014 October release)] from East Asian, South Asian, and all-population databases. Deleterious effects of the variants were supported by multiple lines of computational evidence including from SIFT, PolyPhen-2, Mutation Assessor, Mutation Taster, FATHMM, and GERP++. Further information is available elsewhere.10
Phenotype assessment
Clinical, laboratory, and pathologic data were obtained retrospectively by reviewing medical records. Clinical information included assessments of age at symptom onset, muscle impairments, respiratory distress, limitations of extraocular muscles, facial deformity, winged scapula, scoliosis, joint contractures, and physical disability. Laboratory analyses included the serum creatine kinase (CK) level. Muscle imaging with CT fat measurements was available for one patient.
Pathologic assessment
Muscle biopsies were performed in three patients, and one patient had been examined previously at other hospital. Three muscle specimens obtained at our center were taken from the biceps brachii (ID185), paraspinal muscle (ID37), and vastus lateralis (ID131). Frozen muscle sections (5-µm thick) obtained from all muscle specimens were stained with hematoxylin and eosin (H&E), modified Gomori trichrome (modified GT), and reduced nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-tr). Muscle specimens were also analyzed by immunohistochemistry using antibodies against the C-terminus, rod, and N-terminus of dystrophin (Leica Microsystems, Newcastle upon Tyne, UK), dysferlin (Leica Microsystems), α-sarcoglycan (Leica Microsystems), β-sarcoglycan (Leica Microsystems), γ-sarcoglycan (Leica Microsystems), δ-sarcoglycan (Leica Microsystems), α-dystroglycan (Millipore, Billerica, MA, USA), and caveolin (BD Biosciences, San Diego, CA, USA).
RESULTS
Mutational analysis
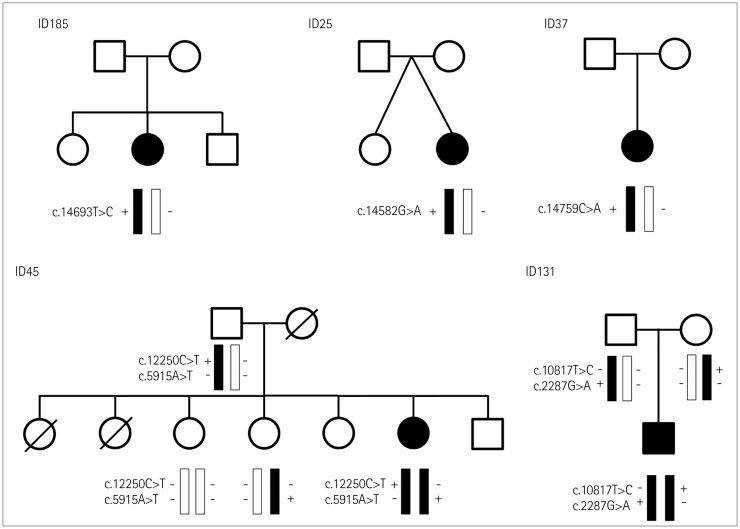
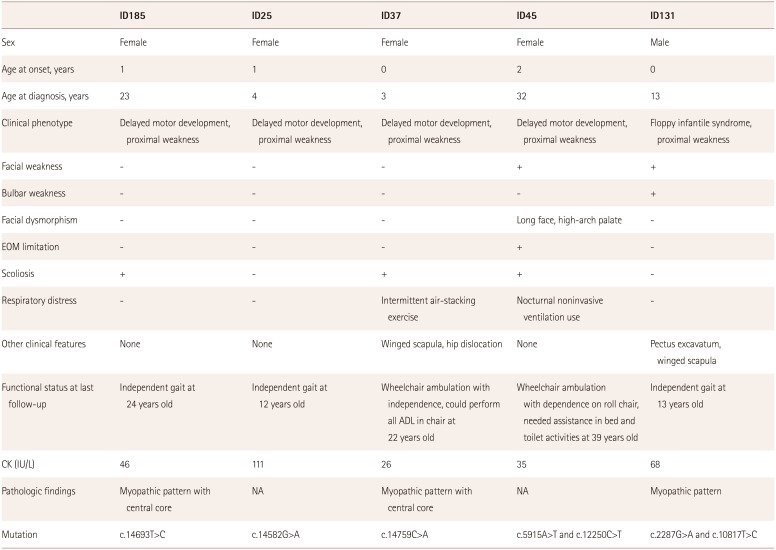
We identified seven different RYR1 mutations in five patients from unrelated families, which were classified into pathogenic or likely pathogenic according to ACMG/AMP guidelines (Fig. 1).11 All of the RYR1 variants that were pathogenic or likely pathogenic were missense variants, and they included two novel variants: c.5915A>T and c.12250C>T. Four patients (ID25, ID37, ID131, and ID185) carried previously reported pathogenic variants,121314 among which ID131 carried compound heterozygous variants (c.2287G>A and c.10817T>C). The proband's parents were both clinically healthy. Genetic testing revealed that the proband's father carried the heterozygous c.2287G>A variant, while his mother carried the heterozygous c.10817T>C variant.
The other patient (ID45) carried two novel compound heterozygous RYR1 mutations: c.5915A>T and c.12250C>T. Sequencing of RYR1 in the family members revealed that the proband's father was heterozygous for the c.12250C>T variant, while her sister was heterozygous for the c.5915A>T variant. These family members with heterozygous variants were clinically healthy. The two no2018-01-05vel variants (c.12250C>T and c.5915A>T) were classified as likely pathogenic according to ACMG criteria, representing two each in the moderate and supporting categories, respectively. The c.12250C>T variant was included in the CCD-MH hotspots and absent from the control. The c.5915A>T variant was absent from the control and detected in a trans arrangement with the c.12250C>T variant. Deleterious effects of these novel variants were supported by multiple sources of computational evidence. We also considered how the features of patient ID45 were compatible with the phenotype of recessive RYR1-related CM.
Clinical manifestations
The clinical features of the five patients with RYR1 mutations are summarized in Table 1. All of the patients presented at infancy, and delayed motor milestones including sitting, standing, and gait were observed consistently, after which all patients had achieved independent gait. However, two patients (ID37 and ID45) became wheelchair dependent at the ages of about 10 and 25 years, respectively.
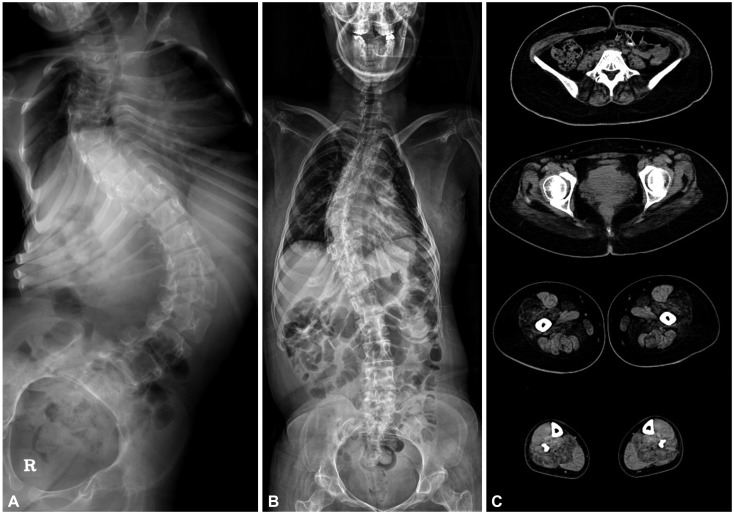
Three patients (ID185, ID25, and ID37) carrying a dominant variant in the C-terminal hotspot demonstrated the clinical phenotype that was previously associated with typical CCD, comprising proximal weakness affecting predominantly the hip girdle, with sparing of extraocular muscles. Two patients (ID37 and ID185) showed scoliosis, among which patient ID37 had severe scoliosis (Fig. 2) with respiratory difficulty and had undergone surgery for scoliosis at the age of 8 years. Although the respiratory function of that patient improved after surgery, she had frequently received interventions for respiratory rehabilitation.
Two patients (ID45 and ID131) carrying compound heterozygous variants demonstrated facial muscle weakness. Patient ID131 presented with floppy infant syndrome, and had pectus excavatum. Although that patient had bulbar muscle weakness, he did not require a nasogastric tube. Patient ID45 showed severe features including facial dysmorphism, scoliosis (Fig. 2), and respiratory distress, and was the only patient to exhibit ophthalmoplegia. This patient experienced respiratory difficulty at 33 years of age, and required nocturnal noninvasive ventilation.
The serum CK level was within normal limits for all patients. None of the five patients had a previous medical history of MH. Muscle CT had been conducted in one patient (ID185), which revealed typical clinical features of CCD and a dominant RYR1 mutation. The results revealed muscle atrophy and fatty infiltration in the lower extremity with a selective involvement pattern–the rectus femoris and adductor longus muscles were spared in the thigh, but the soleus was strongly affected in the calf (Fig. 2)–that was consistent with previous findings for muscle imaging in RYR1-related myopathy.15
Histopathologic examination
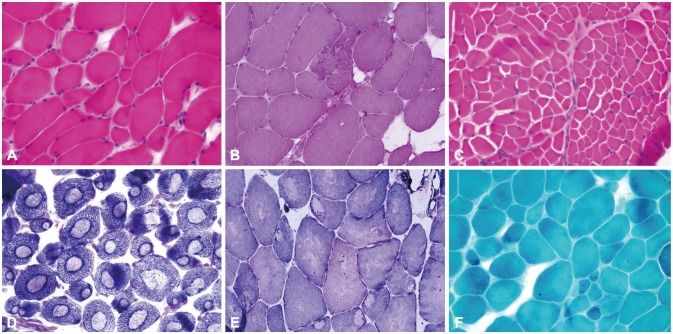
Muscle biopsies were performed in three patients (ID37, ID 185, and ID131). H&E staining demonstrated wide variations in fiber size, degenerative fibers, and increased interstitial fibrosis (Fig. 3A-C). Two muscle specimens from patients ID37 and ID185 showed well-demarcated (Fig. 3D) and ill-defined (Fig. 3E) central cores on NADH-tr staining. Modified GT staining did not demonstrate any rimmed vacuoles, nemaline rods, ragged red fibers, or other granular materials (Fig. 3F). Immunohistochemistry of the muscle specimens showed normal expression levels of dystrophin, sarcoglycan (α, β, γ, and δ), dysferlin, α-dystroglycan, and caveolin in all three patients.
DISCUSSION
The present study identified seven pathogenic or likely pathogenic variants in five Korean patients. We believe that these gene variants were the underlying cause of myopathy in all five patients. Four patients carried previously reported pathogenic variants, while patient ID45 carried two novel compound heterozygous RYR1 variants: c.5915A>T and c.12250C>T.
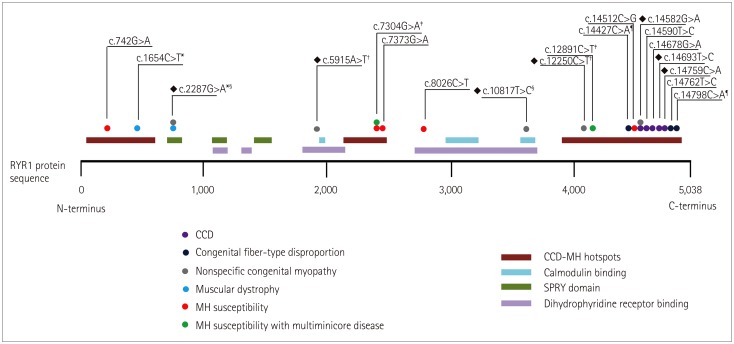
There have been a few reports on RYR1 mutations in Korea. Fig. 4 summarizes the variants reported as pathogenic or likely pathogenic; all of the reported variants are missense variants including two variants in a cis arrangement. These two variants had been reported in an MH family with multiminicore myopathy.16 Most of the patients with MH susceptibility carried a dominant mutation. The reported phenotypes of RYR1-related myopathies comprised three cases of CCD, two cases of CFTD, and one case of muscular dystrophy. The patients with CCD and one patient with CFTD had a dominant mutation in the C-terminal hotspot. The other patient with CFTD and a patient with muscular dystrophy had a compound heterozygous mutation.1417181920
Dominant variants (c.14693T>C, c.14582G>A, and c.14759C>A) in the C-terminal hotspot of our patients were reported previously as being related to CCD. The c.14582G>A (p. R4861H) and c.14693T>C (p.I4898T) variants have been reported in a CCD cohort from European countries.21 In addition, all of the dominant variants (c.14693T>C, c.14582G>A, and c.14759C>A) have been reported previously in a Japanese cohort that showed muscle pathology characterized by the presence of distinct cores in almost all type-1 fibers, and type-2 fiber deficiency.12
Three of the present patients (ID185, ID25, and ID37) carrying dominant variants (c.14693T>C, c.14582G>A, and c.14759C>A) in the C-terminal hotspot that presented during infancy showed proximal dominant weakness and delayed motor milestones. In terms of skeletal manifestations, two patients (ID185 and ID37) carrying variants c.14693T>C and c.14759C>A showed scoliosis, with patient ID37 exhibiting accompanying respiratory impairment and having undergone scoliosis surgery. That patient required wheelchair ambulation after the surgery, which contrasts with a prior report of all patients who received scoliosis surgery maintaining their previous motor abilities.6 Skeletal manifestations were commonly (50–70%) accompanied by weakness in a cohort with RYR1-related CM. In these cohorts, patients with the c.14582G>A and c.14693T>C variants showed skeletal abnormalities including contracture, dislocation, scoliosis, and lordosis.1222 When compared with our cases, a Japanese patient carrying the c.14693T>C variant demonstrated moresevere features than in our case, including floppy infant syndrome, respiratory insufficiency, and generalized weakness. In contrast, another Japanese patient carrying the c.14759C>A variant showed no skeletal abnormality and milder features than in our case.12 Similarly, a previous Korean study of RYR1-related CCD patients carrying dominant variants including the C-terminal hotspot found mild features presenting at childhood without skeletal deformity.19
Advances in the understanding of novel pathogenic mechanisms, in particular underlying recessive RYR1-related myopathies, have expanded the phenotypical spectrum. According to previous studies, patients with CM associated with recessive RYR1 mutations have a tendency for earlier and moresevere presentations compared to patients with dominant mutations.23 In accordance with previous reports, our cases (ID45 and ID131) with recessive variants demonstrated severe presentations.
Among recessive variants, the c.10817T>C (p.L3606P) variant was detected in a trans arrangement with the c.178G>A (p.D60N) variant in a Japanese CCD cohort.12 The reported case had presented at infancy with proximal dominant weakness, delayed motor milestones, and lordosis.12 In contrast, our case carrying the c.10817T>C (p.L3606P) and c.2287G>A (p.V763M) variants in a trans arrangement tended to have an earlier and more-severe presentation compared to the Japanese patient–presenting with floppy infantile syndrome, facial weakness, winged scapulae, and pectus excavatum. Regarding another variant, c.2287G>A (p.V763M) was reported in a trans arrangement with c.1654C>T (p.A552T) in a Korean muscular dystrophy cohort. The clinical features of that patient included weakness, ankle contracture from infancy, and scoliosis.14
The clinical phenotype of our patient carrying two novel variants (c.5915A>T and c.12250C>T) was more severe than those of other patients, and included ophthalmoplegia. According to previous reports of recessive mutations, ophthalmoplegia was reported more frequently in patients who had at least one hypomorphic allele. Conversely, there was no significant association between ophthalmoplegia and mutation position. Regarding pathologic findings, ophthalmoplegia may be associated with internal/central nucleation or a multiminicore-like pathology.923
Two muscle specimens obtained from patients ID37 and ID185 with the C-terminal dominant variant showed core myopathy, which is consistent with common pathologic findings previously reported for RYR1-associated myopathy. Muscle specimens obtained from a patient (ID131) at 3 years of age carrying a recessive RYR1 variant revealed subtle myopathic changes without core. Taking into account that subsequent biopsies showed the gradual development of cores in RYR1-related myopathy,22 we can speculate that this pathologic finding is associated with the age at diagnosis.
It is unfortunate that there is a dearth of information on all patients with respect to MH susceptibility traits such as exertional myalgia, rhabdomyolysis, and in vitro contracture testing, and no study has examined RYR1 protein expression. According to a recent study, exertional rhabdomyolysis or myalgia may be a more-common manifestation of MH-related RYR1 mutations than actual MH itself.24 The huge tetrameric structure of RYR1 makes it highly likely that the genotypephenotypical spectrum will be expanded further. In conclusion, diagnosing and verifying the pathogenicity of genetic variants in RYR1-related myopathies requires a combined approach that integrates clinical, histopathologic, muscle imaging, and genetic findings.
 XML Download
XML Download