

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Guillain-Barré syndrome (GBS) is an acute polyradiculoneuropathy characterized by ascending muscle weakness and areflexia.1 Immune-mediated mechanisms are thought to be responsible for the pathogenesis of GBS. Although this condition has been the subject of a considerable amount of research over several decades, no clear pathophysiological target of human acute inflammatory demyelinating polyradiculoneuropathy (AIDP) has been found. In contrast, a direct causative relationship has been found between acute motor axonal neuropathy (AMAN) and anti-ganglioside antibodies.2 Recent studies have demonstrated the existence of an association between gangliosides and autoimmune mechanisms. Evidence for this association has been gleaned from experimental studies demonstrating the cross-reactivity of autoantibodies induced by ganglioside antigens against the axolemma of the host's peripheral nerves.3,4 Many other studies have demonstrated that this molecular mimicry hypothesis is most consistent with Campylobacter jejuni infection.5
Several of the anti-ganglioside antibodies, including anti-GM1, GM1b, GD1a, and N-acetylgalactosaminyl GD1a (Gal-NAc-GD1a), are common in GBS sufferers from Asian countries and are representative markers of AMAN.5 It was recently revealed that these antibodies are important in determining the electrophysiological characteristics of GBS.6 Uncini et al.7 found that some anti-ganglioside-antibody-positive cases that were initially classified as demyelinating or undetermined types following nerve conduction studies (NCSs) were ultimately revealed to be axonal type on follow-up NCSs. Thus, an accurate classification of GBS subtypes requires serial NCSs. Moreover, this study7 demonstrated that the assay of anti-ganglioside antibodies can be a useful tool for determining the type of GBS at an early stage in the disease.
It is known that diverse clinical features of variant GBS cases can be attributed to each anti-ganglioside antibody.8,9,10,11 For example, the anti-GT1a antibody is the key factor underlying bulbar and brachial palsies in GBS,9,12,13 and the anti-GQ1b antibody is known to be a specific primary factor underlying Miller Fisher syndrome (MFS), and can explain the oculomotor palsy and other cranial-nerve involvement found in GBS.14,15,16 Therefore, investigation of the anti-ganglioside antibodies provides an opportunity to improve the understanding of diverse manifestations of GBS and the related pathomechanisms.5,17 The aim of this study was thus to determine the frequency of anti-ganglioside antibodies in GBS and related clinical syndromes in a Korean population. In addition, the efficacy of conventional electrophysiological study for the diagnosis of AMAN in Koreans was determined.
Methods
Study design
Data were collected from GBS patients admitted to 20 university-based hospitals in Korea. Among the 574 patients who expressed a desire to participate in the anti-ganglioside antibody study for acute peripheral neuropathies during the period of January 2008 to December 2009, 119 clinically compatible GBS cases met the defined criteria and were selected as study subjects.1 Patients with MFS, Bickerstaff's brainstem encephalitis (BBE), or other atypical variants such as a pharyngeal-cervical-brachial (PCB) variant were not included in this study. During the study period, 38, 3, and 5 patients with anti-GQ1b antibody syndrome including classical MFS, BBE, and PCB with positive anti-GT1a antibody, respectively, were encountered. Data regarding the patients' age, sex, type of preceding infection, presenting symptoms, neurological signs, treatment, and cerebrospinal fluid (CSF) findings were analyzed. The GBS disability score, as defined by Hughes et al.,18 was used in this study. Neurological signs were further classified according to the presence of cranial nerve involvements such as oculomotor palsy, facial nerve palsy or oropharyngeal weakness, respiratory disturbances requiring mechanical ventilation, and objective sensory changes.
Anti-ganglioside antibody study
Serum samples were obtained from patients during the acute stage within 2 weeks of symptom onset. An enzyme-linked immunosorbent assay (ELISA) was used to detect the various types of anti-ganglioside antibodies, including immunoglobulin G (IgG) and immunoglobulin M (IgM) antibodies against the gangliosides GM1, GM2, GM3, GD1a, GD1b, GD3, GT1a, GT1b, and GQ1b, as described previously.11 Although they are not true gangliosides, testing was also performed for galactocerebroside and asialo-GM1. The presence and types of anti-ganglioside antibodies were analyzed by researchers who were blinded to the patients' presenting neurological signs and electrophysiological classifications.
Electrophysiological classification
Electrophysiological evaluations were made based on the neurologists' decisions to choose primary axonal form or demyelination when they requested ELISA for anti-ganglioside antibodies.19,20 An initial NCS was performed within 2 weeks of the onset of motor weakness, as described earlier.21 The median, ulnar, peroneal, and tibial nerves were selected for motor NCSs, and the median, ulnar, and sural nerves were selected for sensory NCSs. F-wave evaluations were also conducted from all selected motor nerves. Accordingly, all patients were classified as having primary demyelinating, primary axonal, or unclassified GBS.19,20 Findings of primary axonal or primary demyelinating GBS were further classified as either pure motor or sensorimotor types using electrophysiological parameters. All findings were interpreted by each referring neurology specialist who was blinded to the anti-ganglioside antibody results.
Standard protocol and patient consent
The data for all patients were compiled using a standardized protocol that was reviewed and approved by the ethical committee at the Dong-A University Medical Center, Busan, Korea. Furthermore, informed consent to participate was obtained from all patients or their caregivers.
Statistical analysis
Statistical analysis was performed using Statistical Analysis System (SAS) version 9.0 (SAS Institute Inc., Cary, NC, USA). With respect to the clinical features of the GBS patients, differences in proportions between groups were tested using the chi-square test or Fisher's exact test, and differences in medians were tested using the Mann-Whitney U test. Two-sided tests were used throughout, and the level of statistical significance was set at p<0.05.
Results
Demographics of patients with anti-ganglioside antibodies
In total, 119 GBS patients (diagnosed using the relevant criteria) were analyzed, of which 79 were men and 40 were women. The age at onset ranged from 15 to 80 years (median 55.4 years). Seventy-three patients (61%) had a history of preceding infection, and 60 (50%) were positive for IgG or IgM antibodies against the various types of gangliosides. Of those 60, 58 were positive for IgG-type anti-ganglioside antibodies and 8 were positive for IgM antibodies; thus, 6 of the IgM-positive cases were simultaneously positive for IgG-type anti-ganglioside antibodies.
Analysis of the clinical findings according to the presence of any type of anti-ganglioside antibody revealed that the antibody-positive group had a higher ratio of men, a younger age at onset, more frequent preceding gastrointestinal events with a short interval from infection to motor weakness, less frequent sensory symptoms or signs, and a lower level of CSF proteins (Table 1). Immunomodulating treatment was performed in 87% and 83% of patients from each antibody-positive and -negative group during the acute stage. The modality was intravenous human immunoglobulin in all treated patients, regardless of additional plasmapheresis. Motor disability scores on admission, and at 4 weeks and 3 months thereafter did not differ significantly between the groups (p=0.091, 0.316, and 0.386, respectively).
Clinical significance of each anti-ganglioside antibody
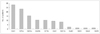
Among the patients who were anti-ganglioside-antibody positive, the most frequent was IgG anti-GM1 antibody (28 patients, 47%), followed by IgG anti-GT1a (23 patients, 38%), and anti-GD1a (15 patients, 25%) antibodies (including overlapping cases) (Fig. 1). Anti-GQ1b antibodies were also found in ten patients, all of whom were also positive for anti-GT1a antibody.
Anti-GM1-antibody-positive cases
Anti-GM1-antibody positivity was strongly correlated with the presence of preceding gastrointestinal infection (61%, p=0.001), negative sensory signs (14%, p<0.001), absence of cranial nerve involvement (25%, p=0.006), and a lower CSF protein level (p=0.004) compared with the antibody-negative group. Motor function score and the frequency of respiratory disturbance requiring mechanical ventilation did not differ significantly from the antibody-negative group.
Anti-GD1a-antibody-positive cases
Patients with anti-GD1a antibody exhibited a similar pattern to the anti-GM1-positive group, with a younger onset age (median 41.3 years, range 25-72 years; p=0.003), frequent occurrences in men (86%, p=0.037), and fewer sensory signs (0%, p<0.001) compared with the antibody-negative group. Anti-GD1a-antibody positivity was frequently associated with facial diplegia, regardless of other accompanying antibodies or the absence of anti-ganglioside antibodies (10/15, 66%; p=0.066).
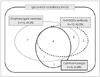
Overlap syndrome of cranial neuropathy associated with anti-GT1a with/without anti-GQ1b antibody
In addition to the typical anti-GM1 antibody related to AMAN pattern, the present results revealed that a large proportion of the patients had anti-GT1a or anti-GQ1b positivity. The IgG anti-GT1a antibody was frequently associated with cranial nerve abnormality (17/23, 74%; p=0.001) (Fig. 2). Compared with the IgG anti-GM1-antibody-positive group, presence of the anti-GT1a antibody was strongly associated with oropharyngeal weakness or other lower cranial neuropathies (16/23, 70% vs. 4/28, 14%; p<0.001). Although ophthalmoplegia appeared to occur more frequently in the anti-GT1a-positive group compared with the anti-GM1-positive group (8/23, 35% vs. 3/27, 11%), the difference was not significant (p=0.084). The simultaneous presence of the IgG anti-GQ1b antibody was more common in the anti-GT1a-positive group than in the anti-GM1-positive group (10/23, 44% vs. 1/28, 4%; p=0.001). Extraocular muscle weakness was strongly correlated with the co-occurrence of anti-GQ1b antibody in the anti-GT1a-positive group (6/10 anti-GQ1b-antibody-positive cases) (Fig. 2). IgG anti-GD1a-antibody positivity was more common in the anti-GT1a-antibody-positive group than in the anti-GM1-antibody-positive group (9/23, 39% vs. 2/28, 7%; p=0.014). Only four cases were simultaneously positive for anti-GM1 and anti-GT1a antibodies, among whom one was also positive for anti-GD1a antibody and another was also positive for anti-GQ1b antibody.
Cases positive for other types of antibody
There was positivity for other minor antibodies against gangliosides GT1b, GD1b, asialo-GM1, GD3, and galactocerebroside in nine, four, three, one, and one case, respectively, regardless of other coexisting anti-ganglioside antibodies. However, there was no clinical significance for these cases. None of the cases were positive for either anti-GM2 or anti-GM3 antibodies. There were no isolated IgG anti-GQ1b-antibody-positive cases.
Correlation between electrophysiological and anti-ganglioside antibody findings
Anti-GM1 and anti-GD1a antibodies are known to be axonal markers, but the patients who were positive for these antibodies did not exhibit significant differences in terms of the proportion of primary axonal pattern at initial NCS in comparison to those who were negative for those antibodies (27% vs. 20%; mean, 7.3 days from symptom onset; p=0.693) (Table 2). All of the diagnoses were made by neurology specialists on the basis of a single, conventional NCS. Twenty-eight of the 41 patients who were positive for IgG anti-GM1 or anti-GD1a antibodies were classified as having primary demyelination at the initial NCS; 21 cases exhibited a pure motor presentation and only 7 patients displayed typical sensorimotor demyelinating polyneuropathy. Conversely, 37 of the 41 antibody-negative patients who were classified as having primary demyelination exhibited a classical sensorimotor polyneuropathy pattern (Table 2).
Discussion
This is the first investigation of anti-ganglioside antibodies and their clinical significance in Korean GBS. The findings show that half of the included Korean GBS patients were positive for various types of anti-ganglioside antibody. The high frequency of anti-ganglioside antibodies revealed herein suggests that they are a continuum of the results obtained in China and Japan.5,20,22 Thus, compared to western countries, the proportion of AMAN seems to be higher in Korea, and similar to those reported in eastern Asian countries. The clinical findings of the antibody-positive group were clearly different from those of the antibody-negative group, although the classes of antibodies detected were diverse. The clinical findings of the antibody-positive group included male predominance, frequent preceding gastrointestinal infection, short interval from infection to motor weakness, negative sensory signs, and lower CSF levels of protein. These findings were also evident when the anti-GM1- or anti-GD1a-positive group was compared with the antibody-negative group. However, a considerable number of the patients who were anti-ganglioside-antibody positive were classified electrophysiologically as having demyelinating GBS. Therefore, the present findings also reaffirm the previous suggestion that using a single electrophysiological test can result in the erroneous misdiagnosis of AMAN as AIDP.7 Overall, it can be assumed that regardless of the electrophysiological classification, half of all Korean GBS patients exhibit the axonal type.
Previous studies have revealed that anti-ganglioside-antibody-positive cases, such as anti-GM1- or anti-GD1a-positive, exhibited different clinical manifestations compared to those with antibody-negative GBS.6,23,24 This finding is attributable to differences in the pathomechanisms of AMAN and AIDP. Although the underlying pathophysiology of AIDP is obscure, damage to the myelin sheath of the sensory and motor nerves by cellular immunity was suggested to occur in AIDP.2 In contrast, several recent studies have provided important information regarding the immunological mechanisms underlying AMAN pathology. According to these studies, the core factor is destruction of the paranodal axolemma induced by humoral autoimmunity, including anti-ganglioside antibodies and the compliment system.25,26,27
One particularly notable finding of the present study was the high frequency of IgG-type anti-GT1a-antibody positivity in Korean GBS. Originally, the anti-GT1a antibody was determined to be a specific marker of the PCB variant of GBS.28 This antibody is closely related to the ophthalmoplegia, facial diplegia, or bulbar palsy.9,29 Since the MFS and PCB variants of GBS were excluded from the present study, the positivity of this antibody in GBS had particular significance with respect to various clinical manifestations. The presence of anti-GT1a antibody resulted in a high proportion of patients with bulbar weakness compared with the anti-GM1-positive group. Anti-GQ1b-antibody positivity within the anti-GT1a group may be responsible for the coexisting ophthalmoplegia; this finding can be explained by cross-reaction with gangliosides of a similar structure.9 Although we do not know the difference from typical MFS cases, one interesting point is that anti-GQ1b-antibody positivity in this series does not appear to be associated with ataxia (only three cases).
Among the different types of cranial neuropathy, facial diplegia was found in 40 patients. The frequency of facial weakness did not differ significantly between the antibody-positive and -negative groups. However, in the antibody-positive group, facial diplegia was strongly associated with the presence of anti-GD1a antibody in this study. Although facial diplegia is a common manifestation of GBS, it can be an isolated sign or a presenting manifestation in some GBS patients.30 Some authors have suggested that prominent facial diplegia is correlated with accompanying anti-GD1a-antibody positivity, although GD1a ganglioside was originally found mainly in the ventral horn and motor fibers of the cauda equina.31,32,33
Electrophysiological evaluation is an important part of understanding the pathomechanism underlying GBS. It is strongly correlated with the presence of anti-ganglioside antibodies, and especially that of GM1, GM1b, GD1a, and GalNAc-GD1a as axonal markers.6 The results of NCSs were compared between anti-GM1- or anti-GD1a-antibody-positive and -negative groups. Unexpectedly, the prevalence of either the axonal or demyelinating subtypes did not differ significantly between these two groups at the initial evaluation according to the NCS criteria.19,20 Previous studies have shown that some AMAN patients exhibit transient conduction blocks in the intermediate and distal nerve segments, mimicking demyelination- a condition known as reversible conduction failure.7,34 This rapidly reversible conduction block, which resolves within days to a few weeks, is found frequently in AMAN patients. This time course suggests functional or microstructural changes at the nodes of Ranvier, rather than segmental demyelination and remyelination; thus, serial NCS is required to confirm the fate of conduction block or axonal degeneration.6,7
The interpretation of the electrophysiological results is limited by only one NCS being conducted in most cases, with no long-term follow up. Although it was not possible to analyze long-term follow-up results, it was noted that the pattern of involvement in the antibody-positive group appeared to be quite different from that in the antibody-negative group. A high proportion of anti-ganglioside-antibody-positive GBS patients exhibited a pure motor type, while most of the antibody-negative GBS patients had typical sensorimotor involvement. It is thus possible that some of the antibody-positive GBS patients could be classified as exhibiting demyelination according to the initial, single NCS criteria, such as prolonged terminal latency or conduction block.6,35,36 Unfortunately, we were unable to analyze the detailed factors contributing to the classification of each case in this study; however, we speculate that the presence of anti-ganglioside antibody and a pure motor presentation on NCS could reflect a different pathophysiological background from that of classical sensorimotor GBS without anti-ganglioside antibodies. In this sense, studies of anti-ganglioside antibodies are important to understanding the various subtypes and manifestations of GBS.
The prognosis did not differ significantly between the anti-ganglioside-antibody-positive and -negative groups. It could be speculated that this is because Korean GBS patients have the opportunity to be treated with intravenous immunoglobulin in the acute stage of the disease, with the support of the national insurance system. Early treatment could halt the reversible conduction failure and prevent axonal degeneration even in AMAN patients. However, this study was subject to important limitations, such as the follow-up period being too short (3 months) and the interpretation of the electrophysiological characteristics being limited. However, it was found that a high proportion of Korean GBS patients expressed a variety of anti-ganglioside antibodies, which may explain the observed diverse clinical characteristics.


 XML Download
XML Download