

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Drug-induced movement disorders include drug-induced parkinsonism (DIP), tardive dyskinesia (TD), tardive dystonia, akathisia, myoclonus, and tremor. Among these, DIP is the most common movement disorder induced by drugs that affect dopamine receptors.1-3 Since the clinical manifestations of DIP are very similar to those of Parkinson's disease (PD), patients with DIP are frequently misdiagnosed as having PD.1,4 These patients are often prescribed antiparkinsonian drugs unnecessarily for long periods of time, despite recovery being possible simply by discontinuing the offending drugs. Dopamine transporter (DAT) imaging may be used in the differential diagnosis of various etiologies of parkinsonism, including DIP.5,6 The aim of this review was to provide clinicians with updated information about the clinical characteristics and DAT imaging findings of patients with DIP, and about the correct treatment for DIP.
Epidemiology of Drug-Induced Parkinsonism
The exact prevalence and incidence of DIP are unclear because it is frequently unrecognized or misdiagnosed as PD. The first study of the extrapyramidal side effects (EPS) of the antipsychotic chlorpromazine found that about 40% of these patients exhibited parkinsonism,7 and several subsequent epidemiological studies found that DIP is the second most common etiology of parkinsonism.8-10 A community-based survey and a population-based study found DIP prevalence rates of 2.7% and 1.7%, respectively, whereas those of PD were 3.3% and 4.5%, respectively.8-10 However, 6.8% of the patients diagnosed with PD were later reclassified as having DIP, thus emphasizing the difficulties in accurately diagnosing DIP and in measuring its prevalence.4 Age is the most obvious risk factor for DIP,11-13 since dopamine concentrations decrease and nigral cells degenerate with age.14 Another risk factor is female gender,8,15-17 suggesting that estrogen suppresses the expression of dopamine receptors;18 however, the exact mechanism underlying the gender difference in DIP remains to be clarified. Genetic factors are also thought to be involved in the development of DIP because not all patients taking dopamine receptor blocking agents (DRBAs) develop parkinsonism.19-21 Genome-wide screening showed that the genes associated with the gamma-aminobutyric acid (GABA) receptor signaling pathway are involved in neuroleptic-induced TD in schizophrenic patients,21 suggesting that genetic factors predispose to both DIP and TD.
Etiology of Drug-Induced Parkinsonism
Typical antipsychotics, also known as neuroleptics, are the most common causes of DIP. However, atypical antipsychotics, which were thought to be free from EPS, can also induce parkinsonism. In addition to antipsychotics, gastrointestinal (GI) motility drugs, calcium channel blockers (CCBs), and antiepileptic drugs have been found to induce DIP (Table 1).
Antipsychotic drugs
The history of DIP parallels that of antipsychotics. Parkinsonism as a side effect of chlorpromazine was first reported 3 years after its introduction.7 It was soon recognized that all typical antipsychotics had the potential to cause EPS, including parkinsonism, acute dystonia, akathisia, and TD.7,22,23 Typical antipsychotics include chlorpromazine, promazine, haloperidol, perphenazine, fluphenazine, and pimozide. About 80% of patients taking typical antipsychotic drugs exhibit more than one kind of EPS.24 Dopamine receptors are widely distributed in the brain, and typical antipsychotics may act on dopamine receptors in the striatum. Therefore, all patients taking antipsychotics have some risk of developing parkinsonism and other EPS. Parkinsonism usually appears days to weeks after starting antipsychotics, but in rare cases the onset delay may be several months or more. The risk of EPS was thought to be low for atypical antipsychotics. Atypical antipsychotics include clozapine, risperidone, olanzapine, quetiapine, and aripiprazole. It was originally thought that their relatively low frequency of associated EPS was due to them being more strongly antagonistic toward serotonin-2A receptors than toward dopamine receptors.25-27 This serotonin-dopamine hypothesis has long been considered a useful model for developing atypical antipsychotics that exhibit superior antipsychotic efficacy with a lower incidence of EPS compared to typical antipsychotics.25-27 The recent 'fast-off' theory suggested that their rapid dissociation from D2 receptors after they have blocked them can explain their lower risk of EPS.28,29
In 1989, clozapine became the first atypical antipsychotic drug to be approved by the US Food and Drug Administration.26 It is effective in schizophrenia patients with drug-resistant negative symptoms, with an almost complete absence of EPS. DIP due to clozapine has not been reported, and it was found to improve psychosis without aggravating parkinsonism even in PD patients.30 However, clozapine has been associated with agranulocytosis in about 1% of patients, making physicians reluctant to prescribe this drug. Other atypical antipsychotics without the risk of agranulocytosis were developed to control psychosis with minimal EPS. Risperidone was expected to have a minimal risk of EPS because it has a high affinity for serotonin receptors.31 However, it binds D2 receptors in a dose-dependent manner, thus inducing parkinsonism and EPS to a similar extent as high doses of typical antipsychotics.31 Although the molecular structure of olanzapine is similar to that of clozapine, it carries with it a significant risk of EPS.32 Quetiapine is an atypical antipsychotic with a low risk of EPS and a low risk of aggravation of parkinsonism when used to treat psychotic symptoms in patients with PD, and is therefore apparently safe for use in elderly patients.33,34 Aripiprazole is the most recently introduced novel atypical antipsychotic, and has a unique mechanism of action. Although it was expected to have a low risk of EPS, clinical experiences have been disappointing.35,36 Thus, to date, only clozapine and quetiapine are associated with low rates of DIP in older patients.
GI motility drugs
GI prokinetic drugs, including metoclopramide, levosulpiride, clebopride, itopride, and domperidone, have also been associated with DIP. These drugs have been used clinically to manage motor disorders of the upper GI tract, including functional dyspepsia and emesis. The prokinetic effect of these drugs is mediated through their blockade of enteric inhibitory D2 receptors.37 Besides binding to receptors in the peripheral end organs, thus inducing antiemetic effects via D2 receptor blockade in the area postrema, they also antagonize central D2 receptors, leading to adverse effects including hyperprolactinemia and EPS. All prokinetics with D2-receptor-antagonizing properties have been found to induce EPS, although the extent of symptoms varies. Among the GI prokinetics, metoclopramide is the most-well-known cause of drug-induced movement disorders.17,38-40 Furthermore, levosulpiride is used widely in several Asian and European countries to treat nausea, vomiting, and functional dyspepsia. Until recently, the drug-induced movement disorders related to levosulpiride were under-recognized, but it has now been shown that levosulpiride frequently causes parkinsonism.41 Whereas metoclopramide usually induces TD, levosulpiride causes parkinsonism more frequently than TD or other EPS. Although metoclopramide and levosulpiride have the same mechanism of action, they show different patterns of adverse effects, the reason for which remains to be clarified. In general, domperidone is considered to be safe for the management of GI discomfort, even in patients with PD, because it does not cross the blood-brain barrier.37 However, although rare, acute dystonic reactions to this drug may occur.42
Other drugs
Parkinsonism may also be caused by CCBs, antiepileptic drugs, and lithium, although their exact pathophysiological mechanisms are not yet known. CCBs including flunarizine and cinnarizine, which are used to control dizziness and headache,43 may cause DIP by reducing dopamine neurotransmission or acting directly on dopamine receptors.44-47 The long-term use of valproic acid was found to induce parkinsonism in 5% of patients;48-50 while the exact underlying pathophysiology has not been clarified, one suggestion is that it is due to oxidative stress and mitochondrial dysfunction.50 Lithium is an infrequent cause of DIP and is thought to act by decreasing dopamine in the striatum, or as an anticholinesterase that increases central cholinergic activity.51,52
Clinical Characteristics of Drug-Induced Parkinsonism
DIP is generally characterized clinically as bilateral and symmetric parkinsonism, with more prominent bradykinesia and rigidity than in patients with PD.13,53 However, it has been suggested in many studies that clinical manifestations alone cannot be used to differentiate DIP from PD (Table 2).
Typically 30-50% of patients with DIP show asymmetric parkinsonism and tremor at rest; these characteristics are considered to support a PD diagnosis.16,41,44,45,54 Interestingly, DIP patients with typical tremor at rest tremors usually also have postural tremors. The similar clinical manifestations of DIP and PD indicate that patients with DIP may have been in a preclinical stage of PD and that their parkinsonism may have been unmasked by the offending drugs. This is supported by findings that parkinsonism persists or even progresses after cessation of the drug in many DIP patients.17,38,40,41,45 In a long-term follow-up study, parkinsonism reappeared several months after the complete resolution from DIP in 7% of patients;41 DIP might have anteceded PD in those patients.
The mean age at symptom onset is greater for DIP than for PD patients. In addition, DIP is more common in females, wh-ereas males are more likely to have PD. While the duration of symptoms is similar, neurological deficits are more severe in DIP than in PD. Orolingual dyskinesia in drug-naïve PD patients is rare, whereas it is frequently combined with parkinsonism in DIP patients. The symmetry and type of tremor are not useful in distinguishing DIP from PD.
Pathophysiology of Drug-Induced Parkinsonism
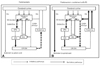
Dopamine receptors in the brain consist of those of the D1 family, comprising D1 and D5 receptors, and the D2 family, comprising D2, D3, and D4 receptors.55 The central dopaminergic system consists of the mesolimbic, mesocortical, tuberoinfundibular, and nigrostriatal pathways. All antipsychotic drugs have potent D2 receptor blocking capacity and the therapeutic effects of these drugs on psychosis are related to their action on the limbic system, where they reduce dopamine transmission. The blockage of D2 receptors by antipsychotic drugs in the striatum leads to disinhibition of GABA- and encephalin-containing striatal neurons at the origin of the indirect pathway without alteration of the direct pathway, followed by disinhibition of the subthalamic nucleus. This leads to increased GABAergic inhibition of the thalamocortical projection by facilitation of the inhibitory projection from the globus pallidus/substantia nigra pars reticulata (Fig. 1A). This pathway resembles the model of disturbance of the basal ganglia-motor loop in PD. More than 80% of D2 receptors were found to be occupied in patients with EPS who were taking neuroleptics,56 in agreement with results showing that clinical symptoms of PD began when over 80% of nigral neurons had degenerated.
TD, defined as hyperkinetic movement in the orolingual or oromandibular area, is caused by long-term use of dopaminergic blocking agents, and frequently accompanies DIP. The co-occurrence of TD with parkinsonism may be due to dopaminergic receptor supersensitivity resulting from long-term D2 receptor blocking. Chronic administration of these drugs increases dopamine D2 receptor density in the striatum. Moreover, withdrawal from neuroleptics was found to aggravate dyskinetic symptoms, whereas increased doses of neuroleptics transiently suppressed dyskinesia. D1 receptors may also be involved in the development of orolingual dyskinesia when D2 receptors are chronically blocked. Chronic administration of D2 receptor blockers also induces changes in the direct pathway of the basal ganglia-motor loop to activate the striatonigral pathway and increase the inhibition of the striatopallidal pathway (Fig. 1B).25,57 This imbalance between direct (D1) and indirect (D2) motor pathways and the resulting alterations in the globus pallidus/substantia nigra pars reticulata complex may lead to hyperkinetic orolingual movements, thus explaining the coexistent and sequential development of parkinsonism and dyskinesia.
Diagnosis of DIP and the Role of DAT Imaging
The clinical diagnostic criteria for DIP are defined as 1) the presence of parkinsonism, 2) no history of parkinsonism before the use of the offending drug, and 3) onset of parkinsonian symptoms during use of the offending drug. Since asymmetrical rest tremors are common in many DIP patients and symptoms persist or progress after cessation of the offending drug, patients clinically diagnosed with DIP may include individuals in the preclinical stage of PD whose symptoms were unmasked by the drug.15,41,58,59
DATs are presynaptic proteins in the membrane on terminals of dopaminergic neurons. They take up dopamine from the synaptic cleft projections that extend from the substantia nigra to the striatum. These transporters control dopaminergic transmission by spatial and temporal buffering, rendering the molecule an imaging target in diseases affecting the dopaminergic nigrostriatal pathway. Single-photon-emission computed tomography (SPECT) and positron-emission tomography (PET) scans are available using several DAT ligands.6,60 SPECT radioligands include 123I-N-3-fluoropropyl-2β-carbomethoxy-3β-(4-iodophenyl)nortropane (123I-FP-CIT or 123I-β-CIT-FP), 123I-ioflupane, DaTSCAN, and 123I-2β-carbomethoxy-3β-(4-iodophenyl)tropane (123I-β-CIT).61 PET scans may be superior to SPECT for imaging DATs, in that the lower energy of positrons provides higher resolution, resulting in better image quality with widespread clinical applications.60 However, most DAT imaging studies, including those in patients with DIP, have utilized SPECT.53,62-64
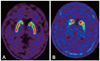
DAT uptake in the striatum is significantly decreased in PD patients, even during the early stages of the disease, because the motor symptoms of PD do not appear until 60-80% of dopaminergic neurons degenerate.65 In addition, drugs causing parkinsonism, such as DRBAs, have negligible affinity for DAT.66,67 DAT scans may show symmetric uptake of radiotracer in the bilateral striatum in patients with pure DIP, even if they have significant parkinsonism (Fig. 2A). PD can be diagnosed in DIP patients whose DAT uptake decreases asymmetrically in the striatum (Fig. 2B). Therefore, DAT scans may be useful for differentiating PD unmasked by drugs from pure DIP. Follow-up DAT scans of DIP patients with initially normal DAT scans exhibited normal uptake in the striatum, whereas scans of patients with initially decreased DAT scans in the striatum exhibited progressive reduction of striatal tracer uptake.62 These findings suggest that DAT imaging provides useful information for diagnosing DIP and may help in the design of a therapeutic plan. Further studies involving larger numbers of patients are needed to determine the sensitivity and specificity of DAT scans in DIP diagnosis.
Treatments and Outcomes of DIP
DIP is generally treated by cessation of the offending drugs. Patients who cannot stop taking antipsychotic drugs because of their psychiatric diseases, such as those with schizophrenia or major depressive disorders, may be switched to atypical antipsychotics that have a lower risk of EPS. People who are prescribed dopamine antagonists due to simple GI disturbance, headache, dizziness, or insomnia should stop taking the offending drugs as soon as possible. Anticholinergics including trihexyphenidyl, benztropine, amantadine, and levodopa have been empirically tested for their ability to relieve symptoms of DIP, but this has produced no clear evidence of their effects in DIP patients.3,16,54,68,69
DIP usually resolves within weeks to months after stopping the offending drug; however, parkinsonism may persist or progress in 10-50% of patients. The prognosis of patients with DIP can be classified into the following types: 1) full and long-lasting recovery from DIP with no subsequent development of parkinsonism, 2) persistence but not progression of parkinsonism, 3) persistence and eventual worsening of parkinsonism, and 4) full remission of parkinsonism but later reappearance after discontinuation of the offending drug. Only patients classified as type 1 can be defined as having 'pure DIP', whereas those classified as type 3 or 4 may be in the preclinical stages of PD.67 The finding of Lewy bodies in two DIP patients who had completely recovered after stopping the offending drugs70 suggests that clinical course alone cannot distinguish 'pure DIP' among patients clinically diagnosed with DIP. In DIP patients classified as type 2, the persistence of parkinsonism may be due to permanent DRBA-induced damage to dopamine receptors.71,72 DAT imaging may be useful in diagnosing DIP patients and predicting their clinical course. A large prospective study may provide more information on the exact prognosis and factors related to recovery in DIP patients.
Conclusions
DIP is important because it is a common etiology of parkinsonism and is frequently either unrecognized or misdiagnosed as PD. In addition, parkinsonism in DIP patients is sufficiently severe to affect daily activities and may persist for long periods of time even after cessation of the offending drug. DAT imaging may be useful for accurately diagnosing patients with DIP and may help to identify the clinical characteristics and exact prognosis of this disorder.
About 50% of patients with DIP and other movement disorders are treated with DRBAs for conditions unrelated to psychosis, including depression, GI disturbance, anxiety, and insomnia.1 Physicians should avoid prescribing DRBAs and CCBs for inappropriate reasons such as anxiety, insomnia, dizziness or dyspepsia in elderly patients and should monitor these patients' neurological signs, especially parkinsonism and other movement disorders, when prescribing these drugs.


 XML Download
XML Download