

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant hereditary disorder characterized by the combined occurrence of tumors of the parathyroid glands, pancreas and anterior pituitary gland [1,2,3]. In addition to these major lesions, adrenal gland tumors, facial angiofibroma, collagenoma, ependymoma, and thyroid lesions are also observed [2,3]. The prevalence of hyperparathyroidism is more than 90% in MEN1 patients and that of pancreas tumors and pituitary tumors are 40% to 70% and 30% to 60%, respectively [3]. In Korean literature, since the first case of MEN1, diagnosed radiologically, was reported in 1986 [4], a total of 46 cases have been reported including the present study. Among total cases, 28 unrelated patients were confirmed by the germline mutations of the MENIN gene, but there is a scarcity of data delineating genotype-phenotype relationships in Korean patients.
MEN1 is caused by germline mutations of the MENIN tumor suppressor gene linked to the chromosomal locus 11q13 [5,6]. The gene consists of one untranslated and nine coding exons, encoding the 610 amino acid protein, menin [5]. More than 400 germline and somatic mutations in MENIN have been found throughout the whole coding sequences, and most of the mutations predict a premature protein truncation [2,5]. However, the absence of a mutational 'hot spot' and lack of genotype-phenotype correlations in MEN1 have been established so far [2,7,8]. This challenging finding might have been affected by additional genetic or epigenetic changes involved in tumorigenesis.
Growing evidence shows that abnormal DNA methylation, along with genetic alterations lead to altered patterns of gene expression in tumorigenesis [9]. DNA methylation plays an important role in silencing tissue-specific genes, imprinted genes, and repetitive elements [10]. Recently, comprehensive analysis of DNA methylation alterations in benign and malignant parathyroid tumors has been reported [9]. However, little is known about tissue specific methylation, especially regarding the MENIN gene in parathyroid tumors due to germline mutation of MENIN.
Therefore, we investigated the results of extensive genetic analysis of MENIN together with clinical phenotypes, and analyzed genotype/phenotype correlations in Korean MEN1 patients. Furthermore, we examined DNA methylation status in MEN1-related parathyroid tumors and compared those to patterns observed in corresponding peripheral blood.
METHODS
Patients
We first reviewed the medical records of 14 unrelated MEN1 patients who had germline mutations of the MENIN gene confirmed in Severance Hospital (Seoul, Korea). We evaluated the extended data from a total of 28 unrelated patients including 14 cases in the present study and 14 cases previously reported with genetic confirmation in Korea by searching on PubMed and KoreaMed [11,12,13,14,15,16,17,18,19,20,21]. The clinical characteristics of six family members were collected for MENIN germline mutation carriers to analyze any relationship between genotypes and phenotypes. The diagnosis of MEN1 was based on the presence of at least two of three main MEN1-related endocrine tumors (parathyroid, pituitary, and pancreas neuroendocrine tumors). Familial MEN1 was defined as one MEN1 case and at least one first-degree relative with at least one of the three main MEN1-related tumors, as previously established [22]. Approval was obtained from the Institutional Review Board of Severance Hospital, Yonsei University College of Medicine in 2011 (4-2011-0613).
Germline mutation analysis of the MENIN gene
Genomic DNA was isolated from peripheral blood leukocytes using a QIAamp DNA Blood Mini Kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer's instructions. The nine exons of the MENIN gene as well as their flanking introns were amplified using primers. Polymerase chain reaction (PCR) was carried out using a thermal cycler (model 9700, Applied Biosystems, Foster City, CA, USA) as follows: 35 cycles of denaturation at 94℃ for 30 seconds, annealing at 60℃ for 30 seconds, and extension at 72℃ for 1 minute. The amplicon was purified using Agencourt AMPure XP (Beckman Coulter Genomics, Danvers, MA, USA). Direct sequencing was performed using the BigDye Terminator Cycle Sequencing Ready Reaction kit on an ABI Prism 3130 genetic analyzer. All variants were confirmed based on the NCBI Single Nucleotide Polymorphism database (http://www.ncbi.nlm.nih.gov/SNP) and Human Gene Mutation Database database (http://www.hgmd.org).
DNA methylation profiling
We used MEN1-related parathyroid tumors to investigate DNA methylation profiling because we could only get the tumor tissues after parathyroidectomy. Six MEN1-related parathyroid tumors from the patients in Table 1 (cases 2-1, 8, 12, 19, 25, and 26) and five of their blood samples (cases 1, 2-1, 8, 12, and 25) were obtained, and consent was obtained from all participants in this study. The sodium bisulfite conversion was performed on 1 mg of genomic DNA for each sample with the EZ DNA kit (Zymo Research, Orange, CA, USA), and 200 ng of the converted DNA was used for PCR amplification. Amplified DNA was hybridized on the Infinium HumanMethylation 450 BeadChip (Illumina, San Diego, CA, USA) following the standard protocols. The hybridized images were processed and intensity data was then extracted after scanning using an Illumina HiScan SQ scanner. β Values ranging from 0 to 1 were calculated for the DNA methylation profile of each candidate. Finally, the candidates were ranked and selected base on the delta-beta (difference between β values) using the Illumina's GenomeStudio Methylation Module, as previously reported [23].
RESULTS
Germline mutations of the MENIN gene in Korea
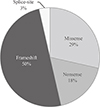
In the present study, we identified 14 germline mutations of the MENIN gene in seven familial and seven sporadic MEN1 patients by direct sequencing. Herein, we extensively collected germline mutations of the MENIN gene in Korean MEN1 patients from previous reports. There were 28 germline mutations in total 34 members of 14 familial (50%) and 14 sporadic cases (50%) (Table 1), and the distribution is shown in Fig. 1. Majority of the mutations were concentrated in exon 2 (11/28, 39%), 9 (4/28, 14%), and exon 10 (3/28, 11%) which are nine known sites of higher frequency previously classified as I to IX, but unexpected higher frequency in the excluded sites of exons 7 and 8 (14%) were also found in our cases (Fig. 1) [3]. Most of the mutations resulted in frameshift changes which were found in 14 unrelated patients (50%), and five nonsense mutations (18%), eight missense mutations (29%), and one splice site mutation (3%) were documented (Fig. 2). In-frame deletion/insertion mutation or large deletion was not found in this study. Six mutations (c.111dupT/p.S38Ffs*79, c.225_226insT/p.T76Yfs*41, c.383_398del16/p.S128Tfs*52, c.746dupT/p.H250Afs*20, c.1150G>T/p.E384*, and c.1508G>A/p.G503N) were newly confirmed as novel in the present study and the four (c.969C>A/p.Y323*, c.973G>C/p.A325P, c.1213C>T/p.Q405*, and c.1049A>T/p.D350V) have been previously described as novel in Korea (Table 1, Fig. 1).
Clinical phenotypes of MEN1 in Korea
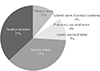
The phenotypic expression of MEN1 in all 34 cases is summarized in Table 1. The most common manifestations were parathyroid tumors (94%, 32/34), pancreas tumors (88%, 30/34), and pituitary tumors (68%, 23/34). Less common MEN1-related tumors were documented in 62% of the patients (21/34): seven adrenal adenomas, six carcinoid tumors (in the stomach, thymus, liver, and lung), two lipomas, and two spinal cord tumors (ependymoma and schwannoma). Finally, 21 patients (62%) had combined tumor involvement of all three main target endocrine glands; 26% (9/34) presented with tumors of the two glands; and 12% (4/34) with one gland.
The first clinical manifestations were available in 30 cases and their mean age was 45 years (median age, 46; range, 21 to 81) (Fig. 3). Three of them (cases 3, 6, and 23 in Table 1) were asymptomatic and incidentally diagnosed at a general health checkup. Two of these cases (cases 3 and 6) had incidentally detected concurrent pancreas and adrenal tumors at the medical checkup. Among 27 symptomatic patients, hyperparathyroidism or hypercalcemia were the first clinical manifestations of MEN1 in 10 patients (median age, 46 years; range 29 to 81 years), pancreas tumors in 10 (median age, 36 years; range 21 to 70 years), and pituitary tumors in three patients (median age, 26 years; range 24 to 34 years) (Fig. 3). Four patients (median age, 44 years; range 27 to 57 years) showed less common tumors associated with MEN1 as a first manifestation, including carcinoid tumors in the stomach or lung and the adrenal cortical adenoma associated with Cushing syndrome (Fig. 3).
Defining the relationship between the germline mutations of MENIN and the associated with clinical manifestations of MEN1, we could not find any direct genotype-phenotype correlations in this study, as already shown by other studies.
DNA methylation profiling in parathyroid tumors
We examined DNA methylation status in MEN1-related parathyroid tumors comparing to those corresponding to the patterns observed in peripheral blood using the Infinium HumanMethylation 450 BeadChip. Distinct hierarchical clustering of genes with altered DNA methylation profiles in blood controls and MEN1 parathyroid tumors was evident (Fig. 4A). The 5' region of MENIN contains a high proportion of "CpG islands." Among these dinucleotides, three sites were significantly hypomethylated in the exon 2 of MENIN: chr11, position 64,577,190 (cg10879244, β=-0.329, P<0.001), 64,577,338 (cg22897141, β=-0.163, P=0.001), and 64,577,427 (cg22527280, β=-0.063, P<0.001) (Fig. 4B).
DISCUSSION
The screening for germline mutations of the MENIN gene makes it possible to perform early diagnosis and better clinical management for mutation carriers. Unfortunately, few studies to date have done genetic testing for MEN1 in Korea; a total of 28 germline mutations (14 familial and 14 sporadic mutations) have been characterized in Korean MEN1 patients. The majority (71%) of the mutations were inactivating ones that would result from the frameshift, nonsenses, and splice site mutations that are consistent with previous reports [2,24,25]. We could not find in-frame deletion/insertion mutations in this work, indicating low frequency of this mutation in Korean MEN1; similar to findings in other populations [2].
Parathyroid tumors are often the first manifestation of MEN1 in more than 85% of patients [26,27]. Meanwhile, hyperparathyroidism by the parathyroid tumor, and pancreas tumors were less common presentations in this study; each appearing in 37% of the patients. On the other hand, 15% of the patients showed unusual initial manifestation of MEN1 (Fig. 3). We identified one case (case 8 in Table 1) with Cushing syndrome due to adrenal cortical adenoma as a first presentation of the disorder. The patient also had spinal cord ependymoma which less commonly develops in MEN1 patients. The prevalence of adrenal lesions in patients with MEN1 varies from 36% to 73% and most of them are nonfunctional [28,29]. Cushing syndrome due to adrenal cortical adenomas as a MEN1-related lesion is very rare, and has been documented in only three cases reported worldwide including this case [30,31]. The other two cases had nonsense mutations (c.781C>T) in the exon 4 [30] or splicing mutation (c.912+1G>A, IVS6+1G>A) in the intron 6 [31]. These mutations would lead to a disruption and loss of function of MENIN.
Four cases (4/35, 11%) had been diagnosed with papillary thyroid cancer concurrently with MEN1-related tumors. Thyroid disease is known to be detected incidentally in over 25% of MEN1 patients [32,33]. Normal thyroid glands have been shown to express menin [34], thus, thyroid tumors in MEN1 patients possibly involved with the MENIN mutations. Genetic analysis of less common tumors such as carcinoids, lipomas, angiofibromas, and esophageal and uterine leiomyomas have reported a loss of heterozygosity (LOH), indicating a role for MENIN mutation in the etiology of these tumors as predicted by the two-hit model of Knudson [35,36]. But no evidence of LOH in the papillary thyroid cancer in MEN1 was reported in two cases [32,36]; thus, it seems that loss of the tumor suppressor function of menin is not required for the tumor formation in these cases. However, in this study, papillary thyroid cancer was the most common manifestation together with adrenal adenoma next to the main three lesions in MEN1 patients. Thus, other genetic alterations without obvious LOH of the gene locus should have occurred.
As we expected, correlations between MENIN mutations and clinical manifestations also appear to be absent in this study. Regarding the two frameshift mutations, c.196_200dupAGCCC and c.200_201insAGCCC (exon 2), each occurred three times, and one, c.628_631delACAG, occurred twice in the different unrelated patients. Even in the patients with same mutations and individual members of the same family, a wide range of MEN1-associated tumors and a lack of genotype/phenotype correlations were revealed. This finding may suggest various clinical manifestations may be caused by tissue-specific modulations such as epigenetic factors. Therefore, we attempted to find epigenetic changes using a methylation study in MEN1-related parathyroid tumors.
In the context of the Knudson two-hit hypothesis, up to 80% of MEN1-associated tumors exhibited LOH of 11q13 [37]. Even though the LOH of parathyroid tumors was not confirmed in this study, we attempted to determine DNA methylation profiles in MEN1-related parathyroid tumors and compare these to respective blood samples. There was altered DNA methylation in the MENIN gene of MEN1-related parathyroid tumors. DNA methylation of the first exon is associated with tissue-specific regulation of gene expression [38]. Although we were not able to determine the role of the three hypomethylations in the MENIN gene identified in this study, these modifications possibly play a role in the parathyroid tumorigenesis and future studies are warranted.
To summarize the status of Korean MEN1: (1) there were relatively high frequency of the mutations in exons 7 and 8 of the MENIN gene; (2) unusual initial manifestations of MEN1 were present in a quite a few MEN1 patients in Korea; thus, genetic testing can provide important information for clearer diagnosis; and (3) the analysis for epigenetic factors regarding tumorigenesis should be considered more carefully.



 XML Download
XML Download