

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hypophosphatemic rickets consists of inherited and acquired forms, which share common pathophysiology and clinical features [1]. In hereditary cases, X-linked hypophosphatemic rickets (XLH) with inactivating mutations of the phosphate regulating gene with homologies to endopeptidases (PHEX) gene is the most common [2]. Autosomal dominant hypophosphatemic rickets and autosomal recessive hypophosphatemic rickets have also been reported [3,4]. Acquired hypophosphatemic rickets includes tumor-induced osteomalacia (TIO) [5] and sporadic hypophosphatemic rickets [6].
PHEX is located on the X chromosome at Xp22.1 [7]. PHEX has been observed in cartilage, bone, and teeth osteoblasts [8] and is thought to act on substrates that suppress phosphate excretion in the proximal renal tubule [9]. Recently, several mutational analyses of the PHEX gene in XLH rickets and sporadic hypophosphatemic rickets reported deletion, insertion, missense, nonsense, and splicing site mutations [10]. Here, we present the first report of a new splicing-site PHEX gene mutation in a Korean patient with sporadic hypophosphatemic rickets.
CASE REPORT
A 50-year-old woman visited the hospital for evaluation of right thigh pain. She had recently suffered from generalized bone pain and generalized weakness, which were aggravated after a fall 6 months prior. Additionally, her waddling gait had worsened. She was born at full term, and her body weight and height were within normal ranges at birth. She had short stature and difficulty in walking when she was a teenager but was not referred to the Department of Pediatrics for evaluation or treatment because she experienced no pain or weakness at that time. She had very short stature (129 cm, just below the 10th percentile for her age) and bowing leg deformities. She was born from healthy parents of normal heights. Her daughter and son have normal stature (her daughter is 160 cm and her son is 180 cm), and the rest of her family is healthy. DNA samples of the family were not available.
She presented with hypophosphatemia 2.0 mg/dL (normal range, 2.5 to 4.7). Her measured serum calcium level was 9.0 mg/dL (normal range, 8.7 to 10.6), alkaline phosphatase was elevated (139 IU/L; normal range, 42 to 98), parathyroid hormone was 45.68 pg/mL (normal range, 10.00 to 65.00), 25-hydroxyvitamin D3 was 11.10 ng/mL (normal range, 10.00 to 30.00), and 1,25-dihydroxyvitamin D3 was 21.6 pg/mL (normal range, 25.1 to 66.1). Nephrologic evaluation demonstrated normal diuresis (1,000 mL/24 hours) and normal renal function (estimated glomerular filtration rate of 95 mL/min/1.73 m2). The tubular reabsorption rate of phosphate was 81% (normal range, 80 to 100). The maximum tubular capacity of phosphate per unit volume of glomerular filtrate was decreased to 0.51 mM (normal range, 0.88 to 1.42 for her age group), which revealed insufficient reabsorption of phosphorus in the kidneys. We also evaluated her serum fibroblast growth factor 23 (FGF23) level using sandwich enzyme-linked immunosorbent assay (ELISA) and found it was 109 RU/mL (normal range, <189 for her age group). The interindividual coefficient of variation (CV) for this ELISA assay is 3.8% to 6.4%, and the intraindividual CV is 2.5% to 6.1%.
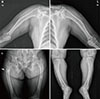
Her plain radiograph presented diffuse bowing deformities in the upper and lower extremities and osteoporotic changes in the extremities, spine, and pelvis. There was a right subtrochanteric fracture that included the cortex of one long bone. If she had preferred surgery, it might have been possible. However, the symptoms were not severe, and she could walk by herself without orthoses. Furthermore, multiple pseudofracture lines were also observed in the right proximal femur, left iliac bone, and proximal shaft of the both fibulas with anterolateral bowing (Fig. 1). Dual energy X-ray absorptiometry measurement showed that the mean bone mineral density in the lumbar spine (L1 to L4) was 0.845 g/cm2, and that in the femoral neck was 0.661 g/cm2, resulting in T-scores of -2.7 and -2.3, respectively. After obtaining informed consent from the patient, genomic DNA was extracted from peripheral blood leukocytes using QuickGene Blood kit (Fujifilm, Tokyo, Japan). For PHEX gene analyses, all 22 exons and exon-intron junctions of the PHEX gene were amplified using polymerase chain reaction (PCR) with the appropriate primers (Table 1). Subsequently sequence analyses were performed with BigDye terminator version 3.1 (Applied Biosystems, Foster City, CA, USA) and the same primers used for PCR amplification. We found a novel splicing mutation in the exon 5-intron 5 boundary of PHEX, a heterozygote of the c.663+1G>A variation, which had not been previously reported (Fig. 2).
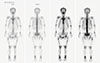
In chromosomal analyses of 20 cells in metaphase which were performed using 72-hour culture and G banding stain of peripheral blood, mosaicism for trisomy X, and tetrasomy X were unusually expressed. We investigated serum tumor markers and whole body bone scintigraphy using Tc-99m hydroxy methylene diphosphonate (Fig. 3) to identify neoplastic lesions and found no evidence of TIO.
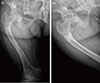
The patient was treated using alfacalcidol and elemental phosphate (Joulie's solution; 10 mL of 4.9 mmol phosphate in 1 mL water) four times a day. After 22 days of outpatient care, her measured serum phosphate level increased to 3.6 mg/dL (normal range, 2.5 to 4.7), alkaline phosphatase decreased to 124 IU/L (normal range, 42 to 98), and her generalized bone pain was decreased to allow daily walking. In addition, follow up X-ray findings of her right femur showed improved fracture union (Fig. 4).
DISCUSSION
XLH is the main cause of hypophosphatemic rickets. The gene that causes XLH was identified on Xp22.1, which was found to be PHEX [7]. Although a physiologically relevant PHEX function has not been established, several recent studies indicate that PHEX inactivates a phosphaturic hormone, such as FGF23, or interacts with membrane proteins that modulate phosphorus activity [5]. FGF23 is a representative phosphatonin that stimulates the renal excretion of phosphate in its full-length state by inhibiting sodium-phosphate transport. Physiologically, FGF23 is inactivated by cleavage at the potential recognition/cleavage sites for the enzymes of the proprotein convertase family (RXXR motif) [11]. Elevated circulating levels of FGF23 were observed in autosomal dominant hypophosphatemic rickets of RXXR motif mutation and also in XLH and TIO [3]. In XLH, an elevated FGF23 level is frequently observed in association with hypophosphatemia. Therefore, some authors have suggested that PHEX controls the inactivation of FGF23, and PHEX gene mutations, such as loss of function mutations, increase the circulating level of FGF23 [12]. In in vitro experiments, a furin-like enzyme cleaved FGF23, not directly mediated by PHEX using the Hyp mouse that is homologous to XLH. However, PHEX inactivation resulted in a tendency to increase FGF23 level in a mouse model. Although FGF23 is not a direct substrate of PHEX, the PHEX gene might be associated with FGF23 expression [13]. In our case, normal level of FGF23 and a lower than normal tubular reabsorption rate of phosphate might contribute to the late onset of rachitic symptoms, including generalized bone pain and muscle weakness, despite PHEX gene mutation. In the renal phosphate wasting state, an elevated FGF23 level is frequently found. Although the relationship of elevated FGF23 with PHEX mutation was not elucidated, most hypophosphatemic rickets present with elevated FGF23. However, Jonsson et al. [12] reported elevated FGF23 levels in 13 of 21 patients. Therefore, not all patients with hypophosphatemic rickets have elevated FGF23. In normal adults and children, the FGF23 levels were definitely normal [12]. Shaikh et al. [14] suggested other circulating factors controlling phosphorous metabolism such as secreted frizzled-related protein (sFRP-4), matrix extracellular phosphorglycoprotein (MEPE), and fibroblast growth factor 7 (FGF7). Especially, MEPE transcripts are increased in Hyp mice, which have poorly mineralizing bone [15]. An in vitro study reported that cathepsin B-dependent cleavage is suppressed by PHEX; therefore, MEPE and FGF have been reported as important phosphatonins affecting phosphate homeostasis and skeletal mineralization [16]. In our case, there is a possibility that another phosphatonin may have led to the resulting rickets phenotype. However, our study is limited in that we did not measure the levels of the other factors like MEPE.
Several studies have identified genotype-phenotype correlations of the PHEX gene in hypophosphatemic rickets. In genotype-phenotype analyses of Korean patients with hypophosphatemic rickets, there was no genotype-phenotype correlation, even though skeletal disease is more severe in C-terminal mutations compared to N-terminal mutations [17]. Remarkably, a novel PHEX mutation was discovered in the C terminus of exon 5 here, but the rachitic symptoms were less severe compared to other hypophosphatemic rickets diagnosed in childhood or adolescence. The patient did not suffer from bone pain or gait disturbance until middle age.
In previous cases, 124 sporadic mutations of PHEX gene have been reported (online PHEX mutation data base; http://www.phexdb.mcgill.ca), and 22 of them are splicing mutations. Table 2 summarizes four splicing mutations in some of the previous cases [6,10,18,19]. As shown in the Table 2, mutation site does not correlate with disease severity. In one case, even though serum FGF23 was elevated to 2,430 pg/mL, the patient had normal height and no bone fractures, and the maximum tubular reabsorptive rate of phosphate decreased to 0.41 mmol/L. In addition, the patient showed a mild phenotype unrelated to FGF23 level, as in our case [10]. Based on these results, we hypothesized that C-terminal splicing mutations of PHEX could represent mild clinical phenotypes.
The case we report is also a sporadic gene mutation case that is expressed by a novel splicing mutation (c.663+1G>A) in the exon 5 and intron 5 junction. The radiologic and laboratory findings of the patient were reviewed to exclude TIO, and this is the first sporadic case reported with a splicing site mutation in exon 5 of PHEX and a normal FGF23 level. Although morphologic changes including short stature, leg bowing, and waddling gait were found at an early age, the rachitic symptoms were initiated by a traumatic event (falling down) in middle age. After treatment with alfacalcidol and elemental phosphate, the rachitic symptoms subsided.
In conclusion, we have identified a novel splicing site mutation in the PHEX gene in a Korean patient with sporadic hypophosphatemic rickets. Our results indicate that the inactivating PHEX mutation contributes to hypophosphatemia, but is not related to FGF23. Showing a novel mutation in exon 5, these data will provide an opportunity for further analyses of the genotype-phenotype association.



 XML Download
XML Download