

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Von Hippel-Lindau (VHL) disease is a hereditary autosomal dominant, multiorgan tumor syndrome caused by a germline mutation in the VHL tumor suppressor gene [1]. Germline mutations in the VHL gene lead to the development of several benign or malignant tumors and cysts in many organ systems [1]. The most common tumors are hemangioblastomas of the retina and central nervous system, clear cell renal cell carcinoma (RCC) and pheochromocytoma [2]. Multiple cysts, serous microcystic adenomas and neuroendocrine tumors of the pancreas have also been reported in patients with VHL disease [3]. More than 50% of subjects with VHL disease have pancreatic lesions including serous cystadenomas, multiple cysts, neuroendocrine tumors, or combined lesions, but pancreatic lesions are generally asymptomatic or associated with only mild symptoms [4]. Diabetes or pancreatitis seems to be relatively rare [3,4]. Clinically, types of VHL disease have been classified on the basis of the risk of pheochromocytoma and RCC [5,6]. VHL type 1 is characterized by a low risk of pheochromocytoma, while VHL type 2 is characterized by a high risk of pheochromocytoma. Type 2 is subdivided into type 2A, which has a low risk of RCC; type 2B, which has a high risk of renal carcinoma; and type 2C, which has a risk of pheochromocytoma but not the other manifestations of VHL disease [5,6]. The VHL gene was mapped by linkage analysis to the short arm of chromosome 3 in 1988 [7]. Several hundred germline mutations in the VHL gene have been reported since VHL gene was isolated in 1993 [8]. The VHL gene is a tumor suppressor ubiquitously expressed in adult tissues. Individuals with VHL disease carry one wild-type VHL allele and one inactivated VHL allele, and tumor or cyst development in VHL disease is linked to somatic inactivation or loss of the remaining wild-type VHL allele. Approximately 15% to 20% of VHL patients have large germline deletions, 27% have missense mutations, and 27% have nonsense or frameshift mutation [9]. In general, VHL mutations are extremely heterogeneous and are distributed throughout the coding sequence, although intragenic missense mutations are rarely seen with the first 50 codons. In total, more than 150 different germline VHL mutations linked to VHL disease have been reported. We hereby report a patient with VHL disease caused by a novel frameshift mutation in the VHL gene and who initially presented with gestational diabetes mellitus (GDM).
METHODS
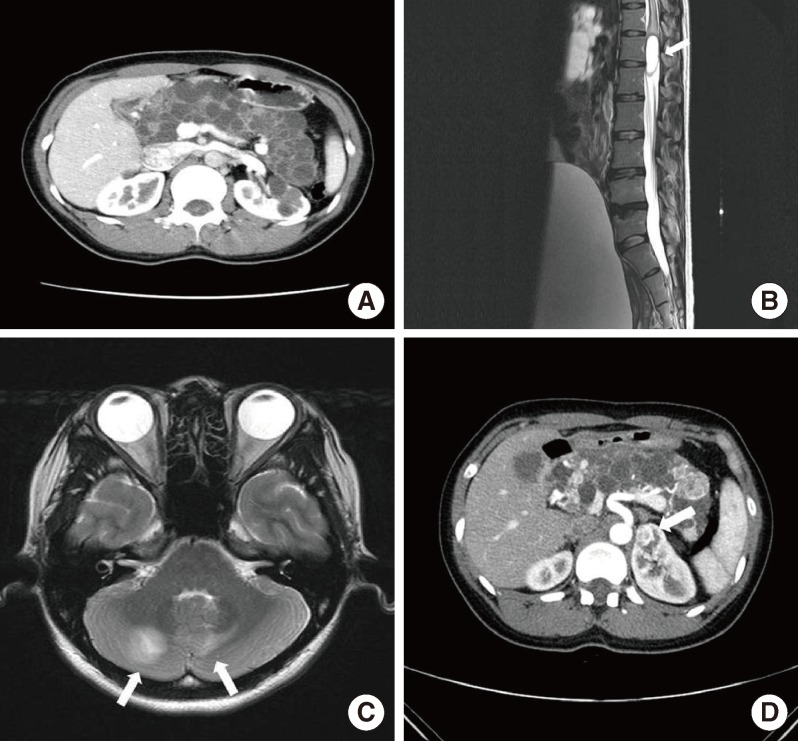
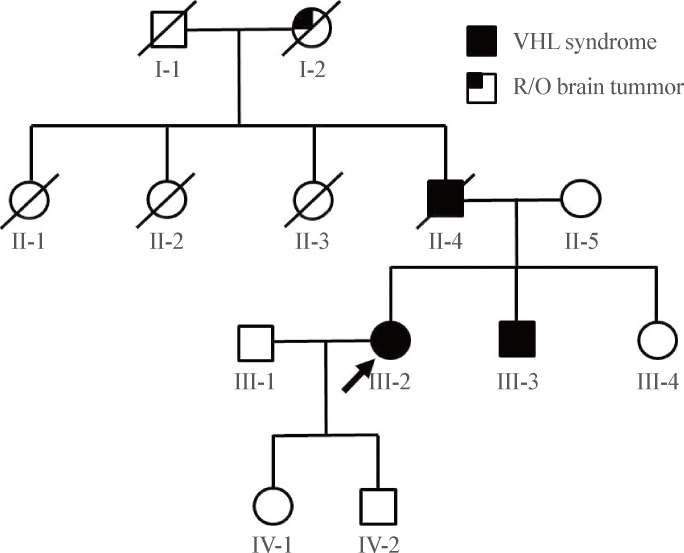
A 30-year-old woman was referred to Seoul National University Hospital diabetes clinic for glycemic control in July 2006. Her height was 159 cm and her body weight was 52 kg, physical examinations including vital signs were within normal limits, and her routine biochemical test results were normal except for the blood glucose level. She was diagnosed with GDM at gestational age 27 weeks of her first pregnancy (in January 2006). She used insulin for glycemic control during the pregnancy. The delivery occurred at full-term without any event, and the baby had no perinatal complications. After delivery, she did not take any antidiabetes medications. At 3 months postpartum, fasting plasma glucose level was 252 mg/dL, 2-hour postprandial glucose level was 572 mg/dL during a 75 g oral glucose tolerance test and her hemoglobin A1c level was 11.0%. Her antiglutamic acid decarboxylase antibody was negative and fasting C-peptide level was 0.2 ng/mL. She needed no more than 10 to 12 units of insulin a day to keep her blood glucose level under control, which was not maintained with oral antidiabetes drugs in the place of insulin. An abdominal computed tomography scan taken in October 2006 revealed that her pancreas and both kidneys were covered with numerous cysts (Fig. 1A); thereby, she was suspected to have VHL disease. Her family members had some features that appeared to be related to VHL disease. Her grandmother was troubled with severe headache during her lifetime, though the cause of death was unclear. Her father, who had diabetes mellitus requiring insulin therapy, underwent brain surgery twice and was diagnosed with brain hemangioblastoma, which was his cause of death. In addition, her younger brother underwent brain surgery in his teens due to brain hemangioblastoma. He had not been diagnosed with diabetes mellitus.
In February 2007, she presented with paresthesia and weakness in both legs and voiding difficulty for 1 month prior to hospital admission. Magnetic resonance imaging (MRI) of the lumbosacral spine showed an intradural, brightly enhancing lesion at the approximated level of the L2 to L3 disc space (Fig. 1B). She underwent complete resection of the tumor. Histopathological examination demonstrated hemangioblastoma. To screen the intracranial lesion, we performed brain MRI. Multiple cerebellar hemangioblastomas were detected on MRI (Fig. 1C), and she underwent gamma knife surgery. Ophthalmologic exam revealed no retinal hemangioblastoma. Two years later (in March 2011), she was diagnosed with RCC and underwent partial nephrectomy on both kidneys (Fig. 1D). Pathology revealed clear cell type RCC.
Genetic analysis of the VHL gene
Informed consent was obtained from the patient and her siblings for analysis of the VHL gene. Peripheral blood samples from the patient, her brother and her sister were collected. The cord blood of the second baby of the patient was obtained at delivery with consent of the patient. A total 50 µL of genomic DNA was extracted using Gentra Puregene Blood Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. To screen the VHL gene for mutations, we performed direct sequencing of the coding region. Each of the three exons of the VHL gene was individually amplified by polymerase chain reaction (PCR), using primer pairs shown in Table 1. After purifying the PCR products by QIAquick PCR Purification Kit (Qiagen) according to the manufacturer's instructions, direct sequencing was performed using an ABI 3730 DNA analyzer (Applied Biosystems, Foster City, CA, USA).
RESULTS
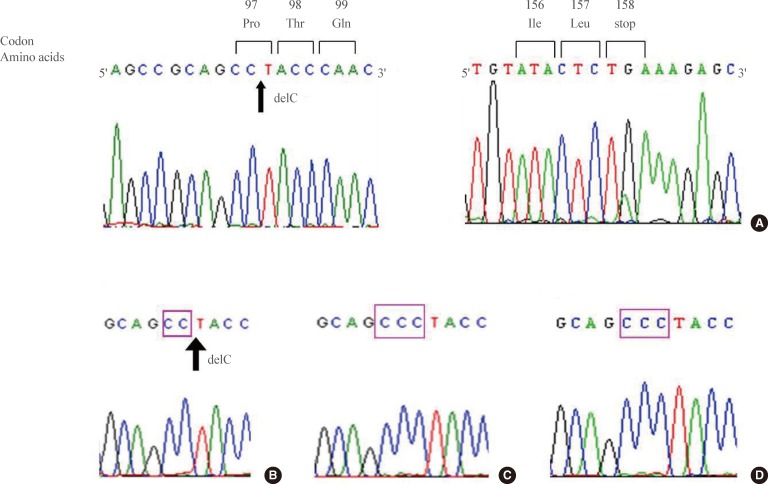
In DNA analysis, we found a 291C deletion mutation in the first exon of the subject's VHL gene (Fig. 2A). This mutation switches the 97th codon from CCC to CCT, which codes for the same amino acid, proline. However, a newly organized 98th codon ACC from TAC produced an amino acid change from tyrosine to threonine (Tyr98Thr). From this point, a frame shift occurred, resulting in the 158th codon acting as an early stop codon (the human VHL consists of 213 amino acids) (Fig. 2A). The human VHL gene is a 12 kb region located at 3p25.3 [5,10]. The gene consists of three exons; the first exon spans 1-113 codons, exon 2 spans 114-154 codons, and exon 3 spans 155-213 codons. Del291C mutation is located in exon 1, and an early stop was introduced in exon 3. Subsequent genetic analysis of family members revealed the same mutation in her brother but not in her clinically unaffected sister or baby (Fig. 2B-D). The assumed genogram based on family history and DNA analysis is shown in Fig. 3.
DISCUSSION
Pancreatic lesions including cysts or neuroendocrine tumors occur in approximately 70% of VHL patients [4,11]. About 90% of pancreatic lesions are single or multiple cysts [12,13]. Serous microcystic adenomas and neuroendocrine tumors of the pancreas have also been reported in patients afflicted with VHL disease. Most pancreatic lesions are asymptomatic and are discovered during screening of family members with VHL disease. However, mass effect of pancreatic lesions can result in acute pancreatitis [4,14], biliary obstruction [4,15] and gastrointestinal obstruction [4,16]. In addition, pancreatic endocrine insufficiency has been reported [4,17,18]. Hammel et al. [4] reported diabetes mellitus to be present in three of 158 VHL patients, and the risk of diabetes was regarded to be relatively low [3,4]. Mukhopadhyay et al. [3] reported that extensive serous microcystic adenomas were associated with diabetes mellitus in patients with VHL disease. Intriguingly, in recent animal studies, loss of VHL tumor suppressor gene in β-cells lead to impaired glucose tolerance in mice [19,20], which indicates that impairment of β-cell function in a patient with VHL disease could be related to an inactivated VHL gene and change of VHL/HIF oxygen-sensing mechanisms.
VHL syndrome is rare in Korea. However, some mutations in the VHL gene have been identified in association with RCC [21], spinal cord hemangioblastoma [22], and pheochromocytoma [23]. Although there have been no large-scale multicenter studies examining the genotype-phenotype correlation, a case series reported that the pancreatic involvement is as high as 89% (16/18) in Korean patients with VHL syndrome [24].
We found a novel mutation (del291C) in the VHL tumor suppressor gene of a patient presenting with GDM. This report suggests that VHL disease can be manifested as GDM as an initial presentation of the disease.

 XML Download
XML Download