

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Newborn screening (NBS) for inherited metabolic diseases (IMDs) is a representative public healthcare project conducted worldwide. In Korea, about 80% of newborn babies undergo NBS for approximately 40 to 50 IMDs. However, only 6 IMDs (congenital hypothyroidism, congenital adrenal hyperplasia, phenylketonuria, maple syrup urine disease, galactosemia, and homocystinuria) have been performed free of charge by the government and other IMDs are not financially covered by the government.1 Traditionally, abnormal NBS result warrants differential diagnosis of the suspected disease, based on an analysis of specific enzymatic or biochemical features in the blood or urine specimens (Fig. 1A). The American College of Medical Genetics and Genomics (ACMG) has developed and established action sheets and algorithms that provide next steps for differentiation of conditions associated with the same or similar metabolic profiles (https://www. ncbi.nlm.nih.gov/books/NBK55827/). However, it is not easy to differentiate IMDs based on biochemical analysis of metabolites alone, which is occasionally impracticable in a clinical setting.2 Furthermore, even different diseases that affect the same metabolic component have distinct and specific treatments and prognoses. In this scenario, a molecular genetic analysis and identification of causative genetic variants are often required to confirm specific disease. Sanger sequencing may require several weeks to distinguish multiple diseases or genes. Therefore, empirical treatment is usually provided while awaiting the final results.3
Recently, several multi-gene panels based on next generation sequencing (NGS) have been developed and validated to facilitate simultaneous and cost-effective analysis of multiple genes related to IMDs or severe primary immunodeficiency. In particular, satisfactory NGS results were obtained with only a small amount of DNA, utilizing minimally invasive procedures with dried blood spots (DBS) or saliva within 1–3 weeks.456
Here, we introduce a new analytical workflow which includes results from both NBS/biochemical assays and targeted gene panel analysis interpreted simultaneously. We demonstrated the efficiency of this workflow for the detection of causative genetic variants in clinically diagnosed patients.
MATERIALS AND METHODS
Study participants and study design
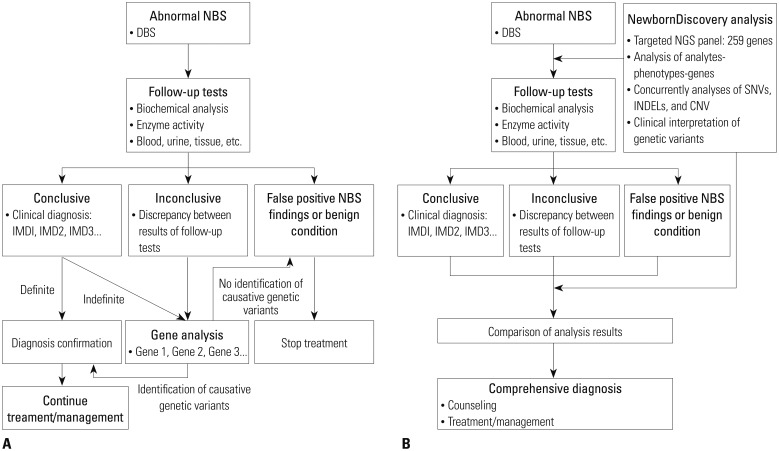
The Institutional Review Boards of Seoul National University Hospital approved and monitored this study (H-1601-079-734). All study participants (parent, legal guardian, and/or child) provided written informed consent to participate in the study. Twenty participants were enrolled into 2 groups (Tables 1 and 2) with each group comprising 10 subjects. No participant had a family history of consanguineous marriage. Group 1 included patients who had been clinically diagnosed with specific IMD based on clinical and biochemical criteria, and were managed prior to enrollment in this study (Table 1). Six of the 10 patients had a type of lysosomal storage disease (LSD) that was not screened by the current NBS in Korea. We analyzed the detection rate of causative genetic variants and the concordance rate between clinical and molecular diagnosis in this group. Group 2 included newborns referred with abnormal NBS tests in an actual clinical setting (Table 2). We performed a pilot prospective clinical study in which this new analytical workflow (NewbornDiscovery, SD Genomics, Seoul, Korea) was applied simultaneously during follow-up standard biochemical tests of plasma and urine metabolites (Fig. 1B).
New analytical workflow for NBS
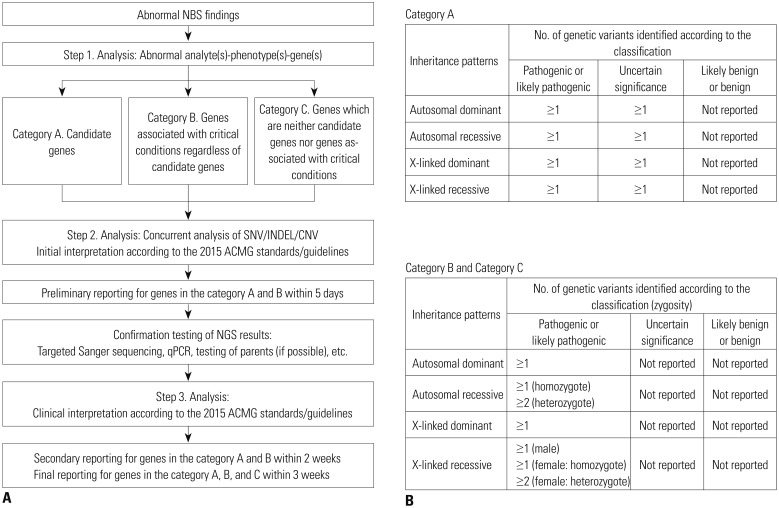
NewbornDiscovery is an integrated workflow design that encompasses concurrent analyte-phenotype-genes (step 1) with single nucleotide variant (SNV)/small insertion and deletion (INDEL)/copy number variation (CNV) analyses (step 2), as well as the clinical interpretation of genetic variants according to the 2015 ACMG standards/guidelines (step 3) (Fig. 2A).7 An NGS panel for NewbornDiscovery comprised 259 genes related to 196 IMDs (including endocrinopathies), 9 hemoglobinopathies, 4 congenital non-hemolytic hyperbilirubinemias, 23 immunodeficiencies, and 17 hearing losses (Supplementary Table 1, only online). After the step 1 (abnormal analyte-phenotype-gene analysis), based on the associated phenotypes, clinical urgency, and incidental findings, NewbornDiscovery categorized the genes as A (candidate genes directly associated with phenotype), B [genes associated with critical conditions according to the 2014 Society for Inherited Metabolic Disorders position statement (http://www.simd.org/Issues/) regardless of candidate genes], or C (neither candidate genes nor genes associated with critical conditions) (Fig. 2A). A classification of identified and reported genetic variants was used differentially according to the gene category (Fig. 2B). In the present study, a preliminary report of genes in categories A and B was generated within 5 days. After additional testing to confirm the NGS results, a secondary report for genes in categories A and B was generated within 2 weeks, and final reporting was completed within 3 weeks.
Analysis and interpretation of variants, and validation of NGS data
A customized panel using a hybridization capture approach was designed to enrich DNA fragments spanning the genes of interest. This panel covered whole coding exons (CDS) and known pathogenic variants in UTR, introns, or promoters in corresponding genes. In order to adequately cover the CDS regions and detect CNV with high accuracy, the probes of all target regions were tiled at a density higher than 3× on average. Genomic DNA was extracted from DBS (total 2 circles measuring 1 cm in diameter; total 4 paper punches with 3.0–3.5 mm in diameter in detail). DNA was isolated on a Chemagic 360-D instrument (PerkinElmer, Baesweiler, Germany), based on magnetic bead technology, using a Chemagic STAR DNA Blood200 kit (PerkinElmer, Waltham, MA, USA) according to the manufacturer's instructions. DNA concentrations were determined using Qubit 3.0 Fluorometer (Invitrogen, Carlsbad, CA, USA). The total genomic DNA yield of each sample was approximately 100–200 ng.
Isolated genomic DNA was fragmented, bar-coded with library adapters, and incubated with oligonucleotide probes. Sequencing was performed with 2×150 bp NextSeq runs (Illumina, San Diego, CA, USA). The NGS quality control matrix was at least 200× mean reads on target with a minimum of 20× coverage on >99.9% target bases. We used an integrated clinical interpretation platform, based on the NewbornDISCOVERY workflow for variant analysis, according to the 2015 ACMG standards/ guidelines.7
The sequence reads were aligned to the human reference genome (GRCh37/hg19) with decoy sequences (hs37d5) using BWA-MEM (version 0.7.12) with the default option.8 The SNVs and INDELs were called using the Genome Analysis Toolkit HaplotypeCaller (version 3.4.46).9 Variant calling was performed with a minimum of 10 reads. Minimum mapping and base quality scores of 20 and 10, respectively, were required to call a variant. The variants were annotated using ANNOVAR.10
CNVs were called using DeviCNV (version 1.0, SD Genomics, manuscript in preparation) developed for exon level CNV detection from targeted gene panel sequencing data, based on read depth analysis. DeviCNV provides a score summarizing quality (confidence intervals of copy number ratios, number of probes supporting the candidates, regression qualities, etc.) for each CNV candidate, and plots for target genes containing copy number ratio with confidence interval of each target probe to help users select clinically relevant CNV candidates for further validation.
To obtain the frequencies of variants in large populations, we used five population databases: the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/), the Exome Variant Server (EVS; http://evs.gs.washington.edu/EVS/), the 1000-Genome Project (1000 GP; http://www.internationalgenome.org/), the Korean Reference Genome Database (KRGDB; http://152.99.75.168/KRGDB/menuPages/intro.jsp), and the dbSNP (https://www.ncbi.nlm.nih.gov/SNP/). Locusspecific databases, such as the ClinVar (https://www.ncbi.nlm. nih.gov/clinvar/), the Human Genome Mutation Database (HGMD; http://www.hgmd.org/), and the Leiden Open Variation Database (LOVD; http://www.lovd.nl/3.0/home), were used to assess the supporting evidence of benign impact or pathogenicity (BP6 or PP5 according to the 2015 ACMG standards/guidelines). To determine possible structural or functional damage due to a missense mutation, we used multiple lines of in silico software such as SIFT,11 PolyPhen-2,12 MutationTaster,13 and FATHMM.14 We also used dbscSNV to predict whether a variant affects alternative splicing.15
We reviewed previously published literature for functional analysis of variants (BS3, PM1, or PS3), population frequencies (BS1 or PS4), or segregation (BS4 or PP1), using an integrated interpretation platform based on expert manual reviews. Finally, we classified variants into pathogenic, likely pathogenic, of uncertain significance, likely benign, or benign categories.
All suspicious SNVs or INDELs associated with patients' disorders were validated with Sanger sequencing. In the case of abrupt decrease in the sequencing depth of specific exons using DeviCNV, we performed exon-based quantitative PCR to detect and validate large deletions containing one or several exons. Quantitative real-time PCR (qPCR) was performed using a QuantStudio 3 real-time PCR system (Applied Biosystems, Foster City, CA, USA). Specific primers were designed for targeted exons and GAPDH (as a control housekeeping gene). All qPCR reactions were performed in a 20 µL final volume with 500 nM each of PCR primers, 2 µL template DNA, and POWER UP SYBR master mix (Applied Biosystems). The qPCR was carried out with an initial denaturation step at 95℃ for 10 min, followed by 40 cycles of denaturation at 95℃ for 15 s and annealing/ extension at 60℃ for 1 min. The segments of the targeted exons and GAPDH were amplified in triplicate for each DNA sample derived from both patient and control subjects. The relative quantity of targeted exons was determined using the 2−ΔΔCt method,16 and the results were normalized against GAPDH as an endogenous control.
RESULTS
NGS analysis
The average reads on target of NewbornDISCOVERY gene panel were 414×. In addition, the NewbornDISCOVERY gene panel yielded 99.9% coverage at 20× coverage and 95.4% coverage at 100× coverage. NGS analysis results are shown in Supplementary Table 2 (only online).
Clinical and biochemical characteristics of Group 1: the clinical diagnosis group
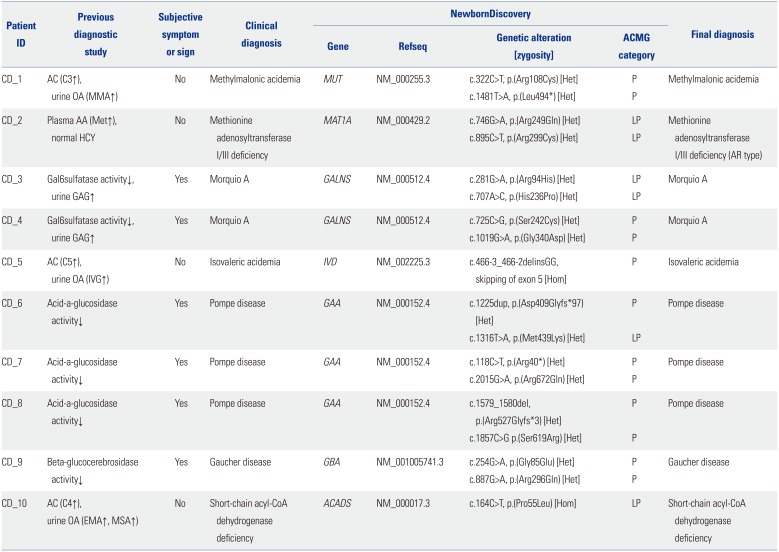
Ten patients in Group 1 were previously diagnosed via biochemical analysis (Table 1). Six of them presented with typical symptoms and signs in accordance with underlying LSDs, three (CD_6–8) with Pompe's disease (OMIM#232300), two (CD_3, 4) with Morquio A disease (OMIM#253000), and one (CD_9) with Gaucher's disease (OMIM#230800). They were clinically diagnosed, based on an analysis of specific enzyme activity and/or pathologic findings of affected tissues at the mean age of 52±49 months (range 8 months–11 years). All patients were undergoing treatment with enzyme replacement therapy at the time of enrollment. The other four patients showed abnormal NBS results without manifesting symptoms or signs, and were clinically diagnosed, based on biochemical studies of plasma and urine metabolites at the mean age of 29±11 days (range 20–42 days). Two patients diagnosed with organic acidemia, CD_1 with methylmalonic acidemia (OMIM#251000) and CD_5 with isovaleric acidemia (IVA, OMIM#243500), were managed with a low protein diet with special milk. CD_2 with methionine adenosyltransferase I/III deficiency (MATD, OMIM#250850) was followed without methionine restriction. CD_10 with short chain acyl-CoA dehydrogenase deficiency (SCADD, OMIM#201470) was managed with riboflavin supplementation together without prolonged fasting. All the 10 patients were followed for 58±28 months and showed normal neuropsychological development.
Molecular genetic analysis of group 1 patients based on NewbornDiscovery
We identified causative genetic variants in all 20 alleles derived from the Group 1 patients (20/20), and the detection rate of NewbornDiscovery in this group was 100% (Table 1). The clinical significance of all identified causative genetic variants was classified as pathogenic (13/20) or likely pathogenic (7/20) according to the 2015 ACMG standards/guidelines.7 No allele in this group was classified as a variant of uncertain significance in the genes under category A.
Eight patients were compound heterozygotes, and all the identified variants were confirmed trans by parental testing. The other two patients were homozygous for either c.466-3_466-2delinsGG on IVD (CD_5) or p.(Pro55Leu) on ACADS (CD_10).
Clinical and biochemical characteristics of group 2 undiagnosed in the pilot study group
All 10 subjects enrolled in this group were newborns with abnormal NBS results (Table 2). The mean age at enrollment was 10.4±5.5 days (range 3–21 days), the mean gestational age was 38.9±1.3 weeks, and no premature infants were enrolled. Their mean birth weight was 3.2±0.4 kg, and all newborns except one (UD_8 with a birth weight of 2.52 kg) showed an appropriate weight for their gestational age. We assessed these patients biochemically using conventional clinical and molecular diagnostic approaches simultaneously according to the protocol indicated in Fig. 1B.
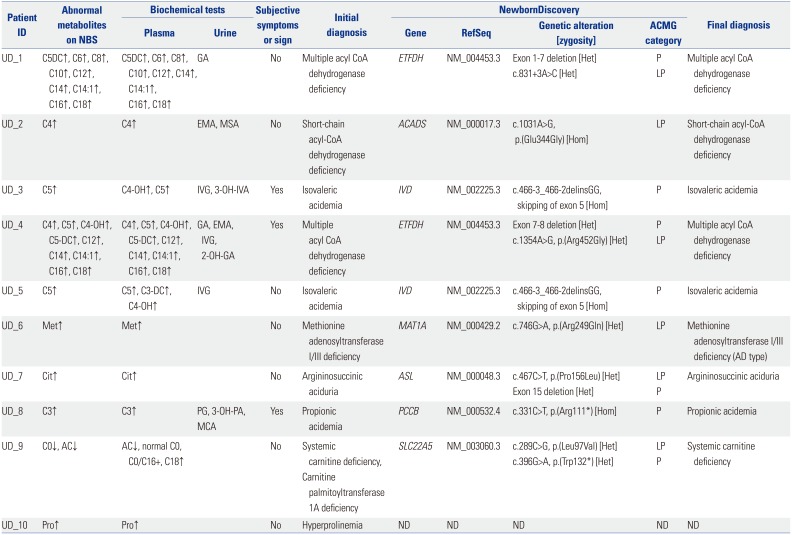
Based on the results of biochemical assays, five patients were diagnosed with possible organic acidemias: UD_8 with propionic academia (OMIM#606054), UD_3 and 5 with IVA, and UD_1 and 4 with multiple acyl-CoA dehydrogenase deficiency (MADD, OMIM#231680). Three patients tested positive for a diagnosis of aminoacidopathies: UD_7 with argininosuccinic aciduria (ASLD, OMIM#207900), UD_6 with MATD, and UD_10 with hyperprolinemia (OMIM#239500, #239510). Two patients were suspected to have fatty acid oxidation disorders: UD_2 with SCADD and UD_9 with systemic carnitine deficiency (SCD, OMIM#212140) or carnitine palmitoyltransferase 1A deficiency (CPT1D, OMIM#255120). In case of UD_9, the findings in the results obtained from initial NBS varied from the follow-up quantitative plasma carnitine profiles. Although this female patient was referred with an abnormally low free carnitine level on NBS, her quantitative plasma carnitine profiles showed a low acylcarnitine level, but normal total and free carnitine levels. These results were inconsistent with the diagnosis of SCD, but suggested the possibility of CPT1D.
Although 7 of 10 newborns showed no subjective symptoms or signs related to specific IMD, 3 newborns presented with clinical symptoms and signs of metabolic crisis even before NBS results were obtained (60 h after birth for UD_3, 18 h for UD_4, and 58 h for UD_8). These 3 patients required neonatal intensive care for 5–17 days, and UD_4 with MADD died at 20 days due to unresolved metabolic acidosis, hyperammonemia, respiratory difficulty, and loss of normal reflexes. We initiated and continued with general and disease-specific management according to presumptive IMDs, based on biochemical findings, in order to prevent the onset or worsening of complications, until confirmation or exclusion of specific IMDs. Except for UD_4 who died in the neonatal period, the other newborns manifested normal developmental milestones without any acute metabolic crisis, although the average duration of follow- up was only 9 to 12 months in this group.
Molecular genetic analysis by NewbornDiscovery of group 2 patients
We found causative genetic variants in 17 of 20 alleles in 9 of 10 Group 2 patients, and the disease confirmation rate of NewbornDiscovery in this group was 90% (Table 2). UD_6 was diagnosed with an autosomal dominant form of MATD carrying a single heterozygous variant. The other eight subjects (UD_1–5, UD_7–9) were diagnosed with autosomal recessive inherited disorders with two independently segregating mutant alleles (1 from each parent). Parental testing revealed 4 variants involving UD_1 (c.831+3A>C in ETFDH), UD_4 [p.(Arg452Gly) in ETFDH], UD_7 [p.(Pro156Leu) in ASL], and UD_9 [p.(Leu- 97Val) in SLC22A5], which were confirmed trans with a pathogenic variant on the other allele. Accordingly, the ACMG categories were adjusted for uncertain significance to likely pathogenic status. Therefore, the clinical significance of all identified causative genetic variants was classified as pathogenic (11/17) or likely pathogenic (6/17).7 Three patients (UD_1, UD_4, and UD_7) showed a deletion allele across one to seven exons of the causing gene, which was confirmed by quantitative PCR. The clinical significance of these CNVs was classified as pathogenic.7
Two IVA patients (UD_3 and 5) in group 2 carried the same homozygous pathogenic variant in IVD, c.466-3_466-2delinsGG, as CD_5 from group 1. Furthermore, the UD_6 harbored a variant, p.(Arg249Gln), which was also identified in CD_2.
There was only one patient (UD_10) who was not molecularly diagnosed via NewbornDiscovery analysis in the present study. Hyperprolinemia type 1 or 2 was suspected based on elevated proline levels (2.1–2.4 fold higher than the cut-off value) in repeated NBSs. However, no meaningful variant in PRODH or ALDH4A1 was detected using NewbornDiscovery, and no suspicious evidence of large deletions affecting both alleles was found from NewbornDiscovery data. Interestingly, the plasma proline levels measured in our hospital showed a tendency to decrease without any abnormality in other metabolic and chemistry studies. The high proline levels on NBS might have been transient and not pathologically correlated.
DISCUSSION
Traditionally, the medical screening protocol for newborns with abnormal NBS is shown in Fig. 1A. Recurrent abnormal results detected on NBS warrant follow-up biochemical tests for diagnosis and management. Follow-up biochemical tests usually include plasma amino acid analysis, urine organic acid analysis, specific enzyme assays, and measurements of toxic metabolites such as ammonia, ketones, and lactic acid. However, discrepancies between the results of biochemical tests have often been observed, and biochemical enzyme assays are labor-intensive and tedious, and yield semiquantitative results.217 In such cases, Sanger sequencing is the next step employed to confirm and differentiate the diagnosis. However, Sanger sequencing is also expensive and is hampered by the genetic heterogeneity of IMDs, which may result in delayed confirmation of diagnosis. In addition, although tandem mass spectrometry (TMS)-based NBS facilitates simultaneous detection of approximately 40–50 metabolites, new therapeutic agents have been developed for the treatment of various IMDs, many of which are not currently included in the NBS lists.18
Multi-gene panels based on NGS techniques represent a potential alternative and complement the current NBS. They are associated with minimal ethical implications related to whole exome/genome sequencing by avoiding secondary findings or findings that cannot be interpreted. NBS should be targeted to diseases that are clinically well defined and medically actionable. Due to possible secondary findings associated with genome-based tests that are independent of the original purpose of NBS, a targeted gene analysis involving multi-gene panel is recommended to integrate NGS-based tests with the NBS.19 The NewbornDISCOVERY analysis is focused on both diagnostic and proactive applications, and provides uncertain to positive results of suspected disorders (in the gene category A) for the diagnostic use. Incidental findings, which are variants in the genes under categories B and C, are reported with clear results for proactive use: 1 or more pathogenic or likely pathogenic variants for autosomal dominant disorders, 1 homozygous pathogenic/likely pathogenic variant or 2 heterozygous pathogenic/likely pathogenic variants for autosomal recessive disorders.
The application of NGS techniques to NBS is technically feasible. Recent studies show that DNA can be successfully extracted from DBS for conventional NBS,46 and one of the studies reported an analytical sensitivity of 99.8%.4 However, the NGS analysis and interpretation are still insufficient and difficult. Above all, the difficulty in interpreting variants of uncertain significance (VUS) should be considered, because the database of genotype-phenotype correlation is insufficient at present. Furthermore, these VUS are detected frequently (Supplementary Table 2, only online). In addition, it is well known that clinical phenotype varies even among family members affected with the same genotype.
The general price of clinical NGS testing (e.g., approximately 500–3000 USD for clinical NGS panel testing) depends on the number of genes analyzed, the range of interpretation or consultation, or inclusion of secondary validation testing. It exceeds 2000 USD for clinical whole exome sequencing and is higher than NBS (approximately 100–200 USD), biochemical assays (approximately 100–500 USD), or targeted Sanger sequencing (approximately 500–1500 USD for whole coding exons) alone. However, if a patient is finally diagnosed with a specific IMD, the new integrated approach (NBS combined with NGS testing) reduces the total diagnostic cost compared with the conventional approach. It is possible to confirm the specific disease earlier, if NGS is implemented at an appropriate stage of the diagnostic process. In addition, it covers most IMDs at low cost, compensates for false-positive results of current NBS, and leads to the identification of new causative genetic variants.4520
In this study, we used a newly introduced analysis workflow, NewbornDiscovery, which comprises 259 genes including those associated with newly treatable or actionable diseases (Supplementary Table 1, only online). In sequence data obtained through multi-gene panel sequencing, many heterozygous variants that are not associated with the patient's actual disease are concurrently identified. Biochemical data are indispensable for the interpretation of the clinical significance of these identified variants and also for the exclusion of or focus on specific diseases. By executing these two different elements of the workflow in parallel and comparing the results of analysis with each other, we not only achieved a higher rate of disease confirmation in a shorter time frame, but also initiated management of a specific IMD earlier and provided accurate genetic counseling to each patient.
In the present study, both patients diagnosed with MADD (UD_1 and 4) harbored different novel sequence variants in an ETFDH allele. The c.831+3A>C from UD_1 was predicted to affect RNA splicing by in silico analyses,15 although analysis of cDNA constructed from the patient's RNA was necessary for confirmation. The p.(Arg452Gly) from UD_4 is a missense variant. In a previous report, a MADD type C patient harbored a variant at the same amino acid, p.(Arg452Lys). However, the clinical significance of this variant has yet to be identified.21 Therefore, based on sequence data alone, these two variants were initially interpreted as VUS.7 However, abnormal biochemical findings to suggest MADD may be considered concurrently using the NewbornDiscovery approach. The NGS data were meticulously investigated focusing on ETFDH, and a large deletion (exons 1–7 for UD_1 and exons 7–8 for UD_4) was quickly found on the other allele using DeviCNV. Finally, the variants c.831+3A>C and p.(Arg452Gly) were re-classified as likely pathogenic after confirmation of their location in trans to the deletion allele by parental testing.7 The p.(Pro156Leu) on ASL from UD_7 was a novel variant, and on another variant in the same amino acid, p.(Pro156Arg) has been reported, in an ASLD patient.22 By identifying a deletion in the other allele using the NewbornDiscovery, we established the molecular diagnosis of ASLD. In total, 3/37 mutant alleles in this study (8.1%) were identified as deletion CNVs.
In the present study, we found additional benefits of NewbornDiscovery, shown in the UD_9 case. The results of the patient's TMS and plasma carnitine profiles were not consistent, and the two suspected conditions, SCD and CPT1D, required different medical management. L-carnitine supplementation is the mainstay of management for SCD,23 whereas CPT1D requires a low-fat and high-carbohydrate diet instead of L-carnitine supplementation.24 Based on NewbornDiscovery, we found two causative genetic variations in SLC22A5 and confirmed the diagnosis of SCD within 2 weeks (and a preliminary report within 5 days), thus facilitating prompt disease-specific treatment for UD_9. The likely pathogenic variant, p.(Leu97Val) located in the first extracellular loop, existed in trans to the pathogenic variant, p.(Trp132*). The Asp91 site near Leu97 is a protein glycosylation site, and normal glycosylation plays an important role in carnitine recognition. Furthermore, a known pathogenic variant near Asp91, p.(Arg83Leu), impairs glycosylation and transfer of the mature protein to the plasma membrane.25
Persistent hypermethioninemia is a unique IMD, characterized by both autosomal dominant and autosomal recessive inheritance depending on the type of causative variants. In the autosomal dominant type, p.(Arg264His) is the most frequently identified variant worldwide,25 whereas p.(Arg249Gln) is commonly found in Korean patients.26
A majority of patients, especially those carrying the autosomal dominant type, show no clinical abnormalities. Mean plasma methionine concentrations show a positive correlation with brain abnormalities on imaging and regular monitoring of plasma methionine levels is recommended.25 In the present study, CD_2 and UD_6 were diagnosed with autosomal recessive and autosomal dominant forms of persistent hypermethioninemia, respectively. The p.(Arg249Gln) was identified in both patients, however, CD_2 carried another recessively inherited variant, p.(Arg299Cys),27 and showed higher methionine levels than those of UD_6. An understanding of the genetic subtypes via NewbornDiscovery facilitates appropriate individual genetic counseling, and prognostic evaluation of the disease.
In the present study, we identified recurrent IMD-related causative variants in Koreans. The c.466-3_466-2delinsGG is the only individual IVD variant found in this study, and all three IVA patients were homozygous for this variant. The c.466-3_466-2delinsGG on IVD was first detected in Korean IVA patients and has been shown to be the most common IVD causative variant identified in Korea.28 Considering that each patient is independent of the others and born out of non-consanguineous marriage, it may represent a founder effect. Two variants associated with ACADS [p.(Pro55Leu) and p.(Glu344Gly)] and detected as homozygotes in CD_10 and UD_2, have also been recurrently identified in Korean and Japanese SCADD patients.293031
In conclusion, the newly developed multi-gene panel, NewbornDiscovery, and the new diagnostic approach combined with biochemical data yielded satisfactory results in both disease confirmation and exclusion. A small amount of blood obtained from a minimally invasive procedure is sufficient to complete the analyses, even in newborns. This approach not only compensates for the disadvantages of current NBS, but also reduces medical costs and socioeconomic losses associated with delayed diagnosis of IMDs. Considering the developmental direction of NBS expansion, we expect that simultaneous testing of TMS and multi-gene panel will become the firsttier NBS in the future.
 XML Download
XML Download