

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
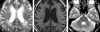
Adult polyglucosan body disease (APBD) was first described in 19711 with at least 49 documented cases found in the literature. This disease is characterized by adult onset (between fifth and seventh decades), progressive sensorimotor or pure motor peripheral neuropathy, upper motor neuron symptoms, neurogenic bladder, and cognitive impairment.1-6 APBD derives its name from the accumulation of rounded, PAS-positive, diastase resistant, intra-axonal inclusion bodies in the central and peripheral nervous system; they are characterized by basophilic hematoxylin-eosin staining and measure up to 70µm in diameter.3,4 Here we describe a case of a 70 year old male patient, review his clinical history, and examine the findings from the neuro-imaging, physiologic studies, and biopsy.
CASE REPORT
A 70 year old man presented with progressive weakness in all extremities, dementia, and constipation about one year ago. Additionally, he complained of bilateral shoulder pain and urinary frequency but did not mention any other urinary symptoms, such as incontinence. He had no family history of neurological disease and no history of seizure or toxin exposure. He was alert, and mental function was preserved on orientation, attention, judgment, immediate and recent memory, but was impaired on delayed recall memory and calculation (MMSE score: 22). Cranial nerve examination was normal. Mild to moderate motor weakness was noted in all extremities (GIII ~ GIV, both proximal & distal). Sensation was intact in all modalities. Deep tendon reflex was decreased; bilateral knee and ankle jerk were nearly absent. Electrodiagnostic studies showed sensorimotor polyneuropathy. A brain MRI showed diffuse white matter and ischemic changes in bilateral deep parietal area, and a focal signal intensity increase in the left dentate nucleus of cerebellum (Fig. 1). Hematological results were normal. ESR was 60mm/hr (normal; 0-10mm/hr), CRP was 63.4mg/L (normal; 0-5.3mg/L). Other serological test results were as follows: ganglioside Ab IgG, HTLV I/II Ab, lupus anticoagulant, anti-phospholipid Ab IgG, IgM, anti-SS-A/Ro, anti-SS-B/Ra, ANA titration, ANA IFA, anti-DNA IFA, anti-HIV I/HIV II, and oligoclonal banding were all negative. The patient's CK level was 6U/L (normal; 35-232U/L), and LD was 352U/L (normal; 225-455U/L). CSF study showed normal findings. Electroencephalography readings were normal. Pathologic study of the left vastus lateralis revealed the presence of polyglucosan bodies in small peripheral nerves of the perimysium, consisting of diastase-resistant glucose polymers when stained with PAS (Fig. 2).
DISCUSSION
APBD should be suspected when patients present with late onset of progressive disease in the peripheral and central nervous system, such as progressive sensorimotor or pure motor peripheral neuropathy, upper motor neuron symptoms, neurogenic bladder, and cognitive impairment.1-6 Its most consistent features are onset within the fifth to seventh decade (88%), peripheral neuropathy (80%), dementia (64%), neurogenic bladder (72%), and upper motor neuron sign (80%), although most patients have one or more of these features missing, and some rarer clinical phenomena have been noted.4 Familial clustering is observed in 30% of cases,3 and the pathogenesis is known as glycogen branching enzyme deficiency.1 Genetic analysis of some Ashkenazi Jewish families and non-Jewish individuals with APBD have identified mutations, but recent reports have also described reduced glycogen branching enzyme activity as an important factor for APBD patients.1 Polyglucosan bodies could be confirmed with a sural nerve biopsy 3,5; some reports showed a skin biopsy also appears to be a simple, reliable, and less invasive diagnostic tool.3 Our patient was confirmed by pathological findings in the peripheral nerves of vastus lateralis muscles. Polyglucosan bodies are also seen in many other diseases such as progressive myoclonic epilepsy (Lafora bodies).3,7 Therefore, typical clinical manifestations, along with histological observation of polyglucosan bodies in nerves, are essential for the diagnosis of APBD.
Herein, we report a case of APBD with typical clinical features and histological findings, the first reported case in Korea.

 XML Download
XML Download