

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Congenital myopathies are a diverse group of skeletal muscle disorders that are genetic in origin, often apparent at birth, are usually slowly or non-progressive, and have overlapping clinical features.1-3 Until recently, the classification of congenital myopathies into distinct groups was most frequently based on the histopathologic features of the muscle.4
Congenital fiber type disproportion myopathy (CFTD) is a congenital myopathy in which the chief pathologic features are small type 1 fibers and type 1 fiber predominance (T1FP).5 Type 1 fibers are considered small when they are more than 12% smaller than type 2 fibers. CFTD is often associated with high arch palate, kyphoscoliosis, contracture, and a slightly increased serum creatine kinase (CK) level.
MATERIALS AND METHODS
Patient selection and muscle biopsy
Three patients with CFTD and six patients with CMT1P were enrolled in this study. The diagnosis of CFTD, a non-progressive neuromuscular disease with predominance and hypotrophy of type 1 fibers, was done clinically and pathologically.5 The diagnosis of CMT1P, a non-progressive neuromuscular disease with predominance of type 1 fibers only, was also done clinically and pathologically.6 Clinical characteristics, serum CK level, cognitive function, electrophysiological studies, and the findings of the muscle biopsy were investigated. The frequency of dysmorphic features such as hip dislocation, kyphoscoliosis, contracture, and high arch palate were also investigated. The Korean Educational Developmental Institute-Wechsler Intelligence Scale for Children (KEDI-WISC) was used for estimation of cognitive function.
All the patients underwent an open biopsy from the vastus lateralis or biceps brachii muscle under local or general anesthesia. Transverse serial frozen muscle sections (10 µm thick) were stained with hematoxylin-eosin (H & E), modified Gomori trichrome, adenosine triphosphatase (ATPase, at ph 9.4, 4.6, and 4.3), and nicotinamide adenine dinucleotide (NADH)-tetrazolium reductase (TR) stains.
RESULTS
Congenital fiber type disproportion (CFTD) group
Details of each patient are given in Table 1. There were 3 male patients (Patients No. 1-3) whose muscle pathology was consistent with CFTD. The age of the patients at muscle biopsy varied from 4 to 5 yr. A nerve conduction study showed normal results in all the patients. A needle electromyography study showed a combination of small brief motor unit action potentials with reduced recruitment in all patients.
One patient (Patient No. 2) had a positive family history. One of his elder sisters had similar symptoms that presented as slowly progressive proximal weakness, scoliosis, and contracture at both ankles over 25 yr. The onset age for these patients varied from 0 to 1.5 yr and the duration of the disease ranged from 3.5 to 29 yr. The onset symptoms included delayed motor milestones in two patients and congenital hypotonia in one patient. Two of the three patients showed facial and neck weakness and respiratory insufficiency. Although all the patients showed a functional disability in walking and running, none were wheelchair-bound at the time of the examination. Two of the three patients showed high arch palate or ankle contracture, scoliosis or lordosis, and right ventricular hypertrophy or T-wave abnormality in EKG. None of the patients showed any hip dislocation. One of the three patients had a low average IQ level. The mean serum CK level was 55 IU/L (range: 34~89 IU/L).
Congenital myopathy with type 1 fiber predominance (CMT1P) group
Details of each patient are given in Table 1. There were six patients (Patients No. 4-9), 4 male and 2 female, who had muscle pathology consistent with CMT1P. The age of the patients at muscle biopsy varied from 0.5 to 5 yr. A nerve conduction study showed normal results in most of the patients. A needle electromyography study showed a combination of small brief motor unit action potentials with reduced recruitment in most of the patients.
None of the patients had a family history or hip dislocation. The onset age for these patients varied from 0 to 6 yr and the duration of the disease ranged from 0.5 to 9 yr. The onset symptoms were delayed motor milestones in 4 patients, congenital hypotonia in one patient, and lower extremity weakness in one patient. One of the 6 patients showed facial and neck weakness, but none of the patients had respiratory insufficiency. One of the 6 patients showed high arch palate, ankle contracture, kyphoscoliosis, but no abnormality in EKG. One of the 6 patients showed delayed development. The mean serum CK level was 155 IU/L (range; 59-261 IU/L).
Pathological findings of muscle biopsy in patients with CFTD and CMT1P
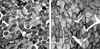
T1FP (> 55%) was present in all 9 patients. Three patients with CFTD had T1FP with type 1 fiber smallness (> 12% smaller than type 2 fiber). The mean percentile of type 1 fiber in CFTD was 73.7% (range: 60-95%). The mean diameter of type 1 fibers in CFTD patients was 43.6 µm and the mean diameter of type 2 fibers was 63.5 µm (Fig. 1A). Six patients with CT1FP showed only T1FP without other structural abnormalities. The mean percentile of type 1 fiber in CT1FP patients was 69.5% (range: 59-90%). The diameter of type 1 fibers in CT1FP patients was similar to that of the type 2 fibers (Fig. 1B). There was no evidence of central cores, minicores, central nuclei, nemaline bodies, reducing bodies, or target fibers in any of the 9 patients.
DISCUSSION
Nearly four dozen congenital myopathies have been described, but the majority of these disorders are rare and considered to be nosologically questionable entities.2 Three firmly established clinical disease entities are central core disease, nemaline myopathy, and centronuclear myopathy. Less common diseases, including congenital fiber type disproportion (CFTD), multicore disease, reducing body myopathy, fingerprint myopathy, and cytoplasmic or inclusion body myopathy, are not as well delineated but are generally accepted as recognizable entities.
T1FP is defined as type 1 fibers composing more than 55% of the total.8 T1FP occurs commonly in conjunction with more specific structural alterations as seen in many of the congenital myopathies.5,9-13
Brook6 and Dubowitz7 introduced the possibility of a distinct entity for children with T1FP without other structural abnormalities found using light or electron microscopy. It has been called "congenital myopathy with type 1 fiber predominance (CMT1P)". Kyriakides et al.14 reported that 9 cases of CMT1P showed hyponia, mild proximal weakness, delayed motor milestones, and normal or mildly elevated serum CK. It is not clear whether "CMT1P" is related to the uniform type 1 fiber entity described by Oh.15 In the present study, four of the 6 patients with CMT1P demonstrated delayed motor milestones and non-progressive proximal lower extremity weakness. One patient showed congenital hypotonia. Another patient (Patient No. 7) showed mild right proximal lower extremity weakness with hip pain. High arched palate, kyphoscoliosis, and contractures were present in one patient. There were no cardiac problems or family histories of neuromuscular disorders. All but one patient (Patient No. 5) with CMT1P showed normal cognitive function. In this study, most patients with CMT1P had mild weakness and normal or slightly elevated CK levels. Considering the clinical, electrophysiological, and morphological findings, the cases lacked distinctive features to support the diagnosis of any other congenital myopathies with specific structural alterations. The diagnosis of hypothyroid myopathy was excluded on laboratory grounds. The clinical manifestations and laboratory findings of these patients with CMT1P were similar to previous reports.14
CFTD is characterized by congenital hypotonia and delayed motor milestones, and is often associated with congenital dislocation of the hip joint, high arched palate, kyphoscoliosis and contractures. Central nervous system abnormalities have been described in some cases of CFTD.16 This disorder has also been associated with hyperinsulinemia and peripheral insulin resistance.17 Electromyographic changes have been different in reported cases of CFTD. In some cases, the electromyography has been described as normal or myopathic but not conclusively diagnostic.18 Other cases have shown neurogenic electromyographic findings such as fibrillation, giant motor unit action potentials, and reduced interference patterns. The serum CK level is normal or mildly elevated. Associated cardiomyopathies have rarely been reported.19 Intelligence is usually normal. The weakness of muscles involved may include those of the legs, arm, trunk, neck or face, but pharyngeal and ocular groups are spared. Despite normal sensation, deep tendon reflexes are usually diminished or absent. In this study, three patients with CFTD demonstrated slowly progressing muscle weakness involving the legs, arm, neck or face, and showed muscle fiber type disproportion in the muscle biopsy. Congenital hypotonia, delayed motor milestones, high arched palate, kyphoscoliosis, and contractures were present in different combinations in this study's patients. Two of the 3 patients with CFTD in this study had cardiac abnormalities. The serum CK level showed normal ranges and the electromyography was described as myopathic in all the patients. The clinical manifestations and laboratory findings of the patients with CFTD were similar to previous reports. The possibility of other congenital myopathies and other diseases, such as congenital myotonic dystrophy, infantile acid maltase deficiency, and Krabbe's disease, was excluded by pathologic, clinical, and laboratory findings.
In this study, the clinical characteristics between CFTD and CMT1P were similar, but the frequency of dysmorphic features was relatively less in CMT1P than in CFTD. Long term observational studies of CMT1P are needed to determine if it will change into another form of congenital myopathy or if CMT1P is a distinct clinical entity.

 XML Download
XML Download