

 PDF
PDF ePub
ePub Citation
Citation Print
Print


A primitive neuroectodermal tumor (PNET) is a malignant, small, round cell tumor that occurs mostly in children or young adults. The most common localization of PNETs is the cerebellum, and is also known as a medulloblastoma. PNETs may also arise in the cerebrum, brainstem, pineal gland, spinal cord, and peripheral nerves (1). Primary spinal PNETs are rare and only 32 cases have been reported in the literature (2345). Primary spinal PNETs can arise as intramedullary, extramedullary, or extradural tumors at any level of the spinal cord. Most primary spinal PNETs are intramedullary and only 8 patients have been reported to have primary spinal PNETs in an epidural location. Here we present an extremely rare case of a primary peripheral PNET confined to the epidural space of the lumbar spine with imaging and immunopathologic findings.
Case Report
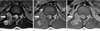
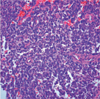
A 12-year-old boy presented to the hospital complaining of lower back pain radiating to both lower extremities. His lower back pain first occurred approximately 1 month earlier and became worse 2 weeks before admission. The neurologic and general physical examination at the time of presentation revealed no specific findings. Lumbar spinal magnetic resonance (MR) imaging revealed an extradural mass with a multi-lobulating contour, extending from the L1 to L2 level with spinal cord compression and extending to the right neural foraminal zone and paraspinal muscle. The right neural foramina was widened and remodeled by the tumor. The tumor exhibited hyperintensity to isointensity with heterogeneity on T2-weighted images (Fig. 1A), isointensity on T1-weighted images (Fig. 1B), and mild enhancement following gadolinium administration (Fig. 1C) with a diameter of 5.2 and 4.1 cm in the axial and sagittal directions, respectively. The patient underwent surgical resection of the tumor with a right hemi-laminectomy and medial facetectomy at the L1 and L2 levels. An extradural-type tumor was noted, which was soft, fragile, and dark gray in color. The histologic examination showed small round cells with rare Homer-Wright rosettes (Fig. 2). Immunostaining showed diffuse and strong membranous staining for CD99 (Fig. 3), and focal staining for synaptophysin, neuron-specific enolase, and vimentin. This immunoprofile confirmed a PNET.
After surgery, the symptoms resolved. Post-operative MR imaging showed no primary lesions elsewhere. Following surgery, the patient received two cycles of chemotherapy (vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide) and one cycle of radiotherapy (4500 rads for 5 weeks). Sixteen months post-operatively, follow-up MR imaging and positron emission tomography revealed no remnants or other tumors. The patient was stable without symptoms at the latest follow-up examination.
Discussion
PNETs have been a controversial subject for more than a decade. PNETs were first described by Hart and Earle (6) in 1973 and included a diverse group of malignant primitive and/or undifferentiated tumors occurring in children and young adults. Initially, PNETs were predominantly undifferentiated tumors of the cerebrum (6); subsequently, central (cPNET) and peripheral types (pPNET) were proposed (7). cPNETs and pPNETs are considered to be clinicopathologically and genetically distinct. pPNETs originate in the soft tissues or bones outside the central or sympathetic nervous system and consist of undifferentiated, small, round-celled tumors with hyperchromatic nuclei and features of neural differentiation with Homer-Wright rosette formation (8). Because pPNETs are classified as small, round-cell tumors, pPNETs must be distinguished from neuroblastomas, non-Hodgkin's lymphomas, rhabdomyosarocomas, and Ewing's sarcomas, which appear to have a strong histogenic relationship. Both pPNETs and Ewing's sarcomas strongly express the glycoprotein, p30/32 (CD99), which is encoded by the microneme protein 2 (MIC2) gene (8). Despite the histologic resemblance, a classification scheme has been proposed for the differential diagnosis of pPNETs and Ewings's sarcomas based of the recognition of neural differentiation characterized histologically by the presence of Homer-Wright rosettes (8). In this case, the immunohistologic examination showed strong immunoreactivies for MIC2 with the presence of Homer-Wright rosettes, which may be diagnosed as a pPNET.
Primary spinal PNETs are rare; only 32 cases have been previously reported (2345). Primary spinal PNETs tend to occur in children and adolescents, with a male predominance. Primary spinal PNETs may arise from all levels of the spine and can be intramedullary, extramedullary, or extradural in location. Intramedullary tumors are the most common primary spinal PNETs, and might originate from the spinal cord, while extradural tumors might arise from vertebrae, soft tissues, or spinal nerve roots.
PNETs have no specific characteristics on clinical presentation or imaging studies. Most of the patients with PNETs present with back pain, as did our patient. Like other malignant spinal lesions, the relevant medical history is brief. Our patient had lower back pain for 1 month; the other previous reported cases had symptoms for < 4 months (3). MR imaging is very helpful in evaluation of the location and extent of the tumor, but no specific imaging findings have been described, and thus may mimic other spinal tumors (39). Most of the imaging findings are hyperintense on T2-weighted images, but the tumor in our case had heterogenous hyperintensity. Other MRI findings showed isointensity on T1-weighted images, heterogeneous enhancement, and widening of the neural foramina, which were described in a previous study and not specific (3). One of the rare differential diagnoses is an extra-osseous Ewing's sarcoma with epidural extension, which is usually hyperintense on T2-weighted images, isointense to hypointense on T1-weighted images, and exhibits moderate heterogeneous enhancement after gadolinium enhancement (10). As the radiologic findings of epidural malignancies are similar to each other, it is difficult to distinguish the radiologic findings without immunohistologic findings prior to surgical diagnosis.
Most pPNETs are treated with surgical resection and adjuvant therapy with chemotherapy and radiotherapy to improve survival rates. Generally, pPNETs cannot be resected en bloc because pPNETs usually involve adjacent nerve roots, the spinal cord, and vertebrae. The prognosis of pPNETs is poor and some patients have recurrences and metastases (345). Our case had surgical resection with chemotherapy and radiotherapy without evidence of metastasis or other abnormal neurologic symptoms.
The present extremely rare case of a primary epidural pPNET of the lumbar spine illustrates the unexpected occurrence and should be included in the differential diagnoses for patients with spinal tumors.

 XML Download
XML Download