

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Kyphoscoliotic Ehlers-Danlos syndrome (kEDS) is an autosomal recessive connective tissue disorder characterized by early onset, progressive kyphoscoliosis, hypotonia, motor delay, hyperextensible skin, skin fragility, and joint hypermobility. Other features include microcornea, ocular and scleral fragility, Marfanoid habitus, atrophic scars on skin and arterial rupture.1 kEDS was initially defined at the biochemical level, on the basis of a family study in which two sisters had progressive scoliosis, joint laxity and recurrent joint dislocations, microcornea and ocular tissues fragility.23
The identified causes were collagen-modifying enzyme procollagen lysine, 2-oxoglutarate 5-dioxygenase 1(PLOD1 or lysyl-hydroxylase 1) deficiency due mutations in PLOD1 and the designated kyphoscoliotic form of EDS.12 Lysyl hydroxylase deficiency results in underhydroxylation of collagen lysyl residues in -Xaa-Lys-Gly-collagen chains and underglycosylation of hydroxylysyl residues, thereby causing impaired collagen cross-linking with consequent mechanical instability of the affected connective tissues.4 Patients with lysyl hydroxylase 1 deficiency have a markedly increased ratio of urinary lysyl-pyridinoline to hydroxylysyl-pyridinoline (LP/HP) due to underhydroxylation of collagen lysyl residues.5 More than 40 different mutations in the PLOD1 gene have been identified in kEDS.6 The large duplication of exons 10–16 is the most common pathogenic mutation, and point mutations, insertions, deletions, and splicing-site mutations have been reported so far.6 Recently, kEDS caused by biallelic mutations in FKBP14 have been identified.7 Patients with FKBP14-related kEDS display a clinically overlapping phenotype with PLOD1-related kEDS and are distinguished by the presence of myopathy, hearing loss, and normal lysyl hydroxylase enzyme activity.6
In infants with kEDS, severe hypotonia and joint laxity lead to delayed gross motor development. Thus, they are usually suspected as having other neuromuscular diseases, which delays a definite diagnosis of kEDS.8910
We report the cases of two Korean siblings diagnosed as having kEDS with progressive kyphoscoliosis, hypotonia, joint hypermobility and skin hyperextensibility. These are the rare cases of kEDS with PLOD1 variants in Korea. Both siblings had two novel deletion variants in the PLOD1 gene.
CASE DESCRIPTION
On March 26, 2019, a 6-year-old girl, was referred to our department for genetic evaluation of progressive scoliosis. She was the second child of non-consanguineous Korean parents. She was born at 35+3 weeks of gestation by caesarean section because of oligohydroamnios. Her birth weight was 2.7 kg and length was 52 cm. After birth, she was found to have severe hypotonia with a lack of spontaneous hand movement and wrist drop, which both led to upper brachial plexus palsy. Mild kyphosis was also observed. She presented with hyperextensible velvety skin, and easy bruising. As classical Ehlers-Danlos syndrome (EDS) was suspected and she had an elder sister with hyperelastic and fragile skin, DNA analysis of the COL5A1 gene was performed with normal results. The severe hypotonia led to delayed motor development. While undergoing physiotherapy, she began standing without assistance in kyphotic posture at the age of 21 months and could walk without support at age 3 years. On physical examination at age 6 years, her weight and height were 20 kg (25–50th percentile) and 116.8 cm (50–75th percentile), respectively. She presented with progressive kyphoscoliosis, pectus excavatum, protruding abdomen, long and slender fingers and pes planovalgus. She showed slightly dysmorphic features including a prominent high forehead, malar hypoplasia, depressed nasal bridges, low set and prominent ears, minimal prognathism, and short philtrum. She had joint hypermobility (Beighton score 6/9), skin hyperelasticity, and multiple atrophic scars on the forehead, knees and back. She had high myopia and keratoconus. Her mother reported that she had ocular fragility and retinal detachment of the left eye after falling down on her face. A skeletal survey revealed thoracic spine dextroscoliosis with kyphotic accentuation in the thoracolumbar junction and both pes planus and genu varum (Fig. 1A and B). Echocardiography revealed no pathological findings. She is currently wearing an orthopedic brace for scoliosis. At age 6 years 8 months, she could walk unsupported and jump, and, go up the stairs with support. Her intelligence was normal.
Fig. 1
Radiologic findings of spine. (A) Spine X-ray of younger sibling showing mild scoliosis at age the age of 5 months and (B) progressive kyphoscoliosis at age 6 years. (C) Spine X-ray of older sibling showing mild scoliosis at age 9 years.
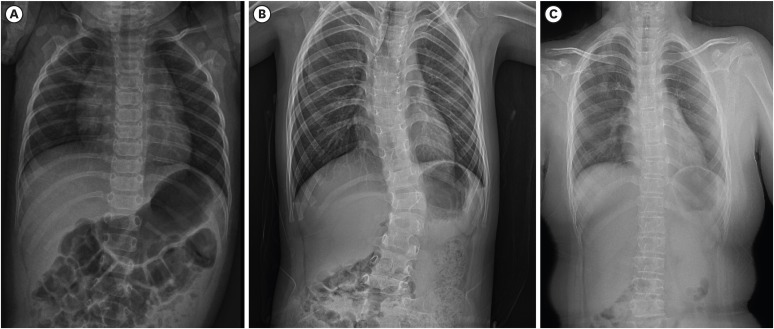
A 9-year-old girl, the older sibling of previous patient, was the first child of nonconsanguineous Korean parents. She was born at 40 weeks of gestation by vaginal delivery. Her birth weight was 2.4 kg. At birth, she had intraventricular and intraparenchymal hemorrhage. She had hyperelastic and fragile skin, easily bruisable skin, and generalized hypermobile joints (Beighton score 6/9). She had high myopia and keratoconus. Her parents reported that she was suspected of having Marfan syndrome due to Marfanoid habitus in infancy. She often fell, which resulted in frequent subluxations of the shoulders and had easily bruisable skin by minor trauma. She was found with marked hypotonia and delayed motor development. She had left hemiparesis due to neonatal intracranial hemorrhage. However, no striking kyphoscoliosis was observed until recently. On physical examination at age 9 years, her height was 143.2 cm (90th percentile) and weight was 48 kg (97th percentile). She showed mild dysmorphic features such as a prominent high forehead, a broad nose, a depressed nasal bridge, low set and prominent ears, and a malar hypoplasia. Multiple atrophic scars were found on the forehead, lower back and knees. Skeletal radiographs revealed a mild scoliosis on thoracolumbar spine and both pes planus (Fig. 1C). The echocardiography findings were normal. The bone density was within the normal ranges. Brain magnetic resonance imaging revealed several oval-shaped high signal lesions in the bilateral periventricular white matter, which suggested sequelae of the periventricular leukomalacia. Currently, she has delayed motor and cognitive development. She walks with support and uses a wheelchair at school.
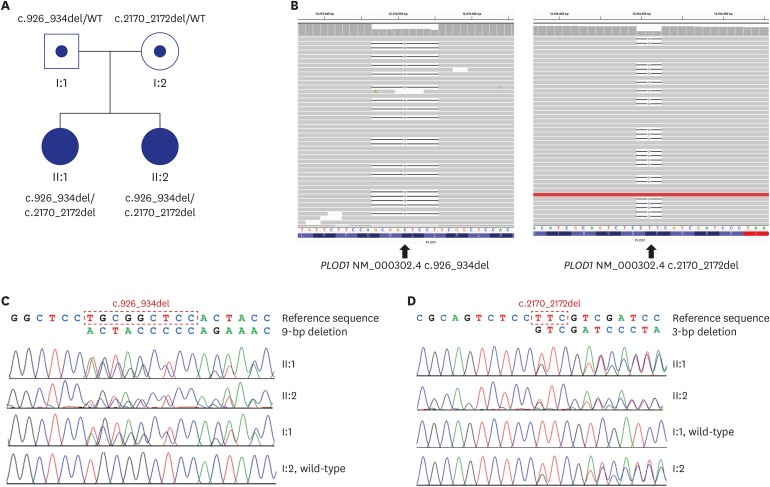
Based on the clinical signs, hereditary connective tissue disorder was suspected. Next-generation sequencing using a targeted panel of 30 genes (ACTA2, ADAMTS10, ADAMTSL4, ATP6V0A2, B4GALT7, CBS, CHST14, COL11A1, COL1A1, COL1A2, COL2A1, COL3A1, COL5A1, COL5A2, COL9A1, COL9A2, ELN, FBLN5, FBN1, FBN2, MYH11, PLOD1, PYCR1, RIN2, SKI, SMAD3, TGFB2, TGFB3, TGFBR1, and TGFBR2) for connective tissue disorders was performed. Genomic DNA was extracted from the peripheral blood of the patient. Library preparation and target enrichment were performed by hybridization capture and massively parallel sequencing was done on the Illumina MiSeqDX (Illuina Inc., San Diego, CA, USA) generating 2 × 150 bp paired-end reads. Sequence reads were aligned to hg19 with Burrow-Wheeler Aligner (version 0.7.10, MEM algorithm). Duplicate reads were removed by using Picard (version 1.138). Local alignment, base quality recalibration, and variant calling was performed with Genome Analysis Tool kit (GATK version 3.5), samtools (version 0.1.19), FreeBayes (version 0.9.21-26-gbfd9832), and Scalpel (version 0.53). Variants were annotated by Variant Effect Predictor and dbNSFP. Common variants were removed with minor allele frequencies ≥1%, according to population databases. The average coverage depth was 230×, and 99.8% of the target bases were covered by more than 10× sequence reads. Two novel variants that were not previously reported in kEDS were identified in PLOD1. The PLOD1 variants were identified in the compound heterozygous state in the two siblings and were validated by Sanger sequencing. Sanger sequencing of their parents revealed that c.926_934del was inherited from the father, resulting in an in-frame deletion of three amino acids leucine-arginine-leucine (p.Leu309_Leu311del). The c.2170_2172del was inherited from the mother, resulting in in-frame deletion of one amino acid phenylalanine (p.Phe724del) (Fig. 2).
Fig. 2
Pedigree and Molecular analyses. (A) Pedigree in the Korean family with kyphoscoliotic Ehlers-Danlos syndrome. (B) Integrative Genomics Viewer snapshot of two novel PLOD1 variants. (C, D) Sanger sequencing confirmation of c.923_934del, inherited from their father (I:1), and c.2170_2172del, inherited from their mother (I:2).
This study was approved by the Institutional Review Board of Soonchunhyang University Bucheon Hospital (SCHBC 2020-02-005). The patient's parents provided written informed consent for publication of patient information.
DISCUSSION
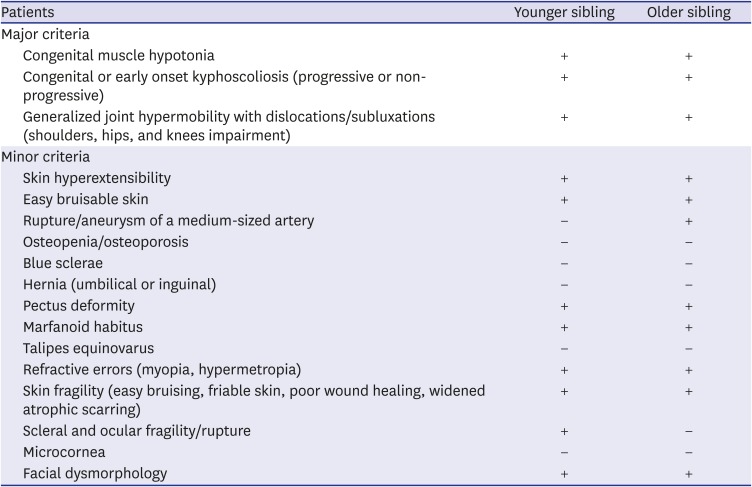
The diagnostic criteria for kEDS were described in the Villefranche criteria in 19981 and revised in 2017.11 The major criteria include congenital muscle hypotonia, congenital or early onset kyphoscoliosis (progressive or non-progressive) and generalized joint hypermobility with dislocations/subluxations (shoulders, hips, and knees in particular). All of our patients met three major criteria and seven minor criteria (Table 1). Moreover, younger sister presented with scleral and ocular fragility, which were rarely reported.36
Table 1
Clinical manifestations of two sisters with kEDS and comparison with revised criteria from 2017 International EDS Classification11
Congenital or early-onset kyphoscoliosis is one of the hallmarks of kEDS. However, some patients with kEDS may develop kyphoscoliosis later in life.412 Without early-onset scoliosis, patients with kEDS may be misdiagnosed as having classical EDS or neuromuscular disorder. In our case, the older sibling who had no congenital or early-onset kyphoscoliosis was thought to have delayed motor development because of intracranial hemorrhage before she was diagnosed as having kEDS. Therefore, if a patient with hypotonia is suspected of having classical EDS but has no COL5A1/2 mutations, the molecular analysis of the PLOD1 gene may be necessary.12 In older sibling, radiographs of the thoracolumbar spine at age 9 years revealed a slight scoliosis, whereas the younger sibling showed kyphoscoliosis in infancy. Only older sibling showed recurrent shoulder joint subluxation. The intrafamilial variation in our case was also observed as previously reported.13 The intrafamilial and interfamilial variabilities of clinical severity and age of onset of kyphoscoliosis in patients with kEDS had no obvious correlation with the PLOD1 genotype.49
Several cases of antenatal or neonatal brain hemorrhage have been reported.4910141516 The neonatal intracranial hemorrhage in older sibling is regarded as the clinical feature of kEDS, although we did not have detailed information about her birth record. In addition, she was thought to have delayed cognitive development because of intracranial hemorrhage caused by birth injury. Vascular events such as aortic dilation/dissection and rupture of medium-sized arteries are the major life-threatening complications, and careful observation and control of blood pressure are necessary to prevent arterial rupture.17
The PLOD1 gene is located on chromosome 1p36.22, is approximately 40 kb in size, and consists of 19 exons.917 The most common mutations, an 8.9-kb intragenic duplication of seven exons (exons 10–16; c.1067_1846dup), is caused by an Alu-Alu recombination in introns 9 and 16, and the allele frequency was 30% in the 42/139 mutations from 73 families.61618 The second most common pathogenic variant in PLOD1 is nonsense variant p.Arg319Ter and the third is p.Tyr511Ter.6 The majority of the others are point mutations, insertion, and deletions and splice site mutations. Two novel variants in PLOD1 were identified in our Korean cases. Two siblings had compound heterozygous variants, c.926_934del (p.Leu309_Leu311del) and c.2170_2172del (p.Phe724del) in the PLOD1 gene and their parents were heterozygous carriers of the variants. These variants were not reported in several global human genome databases, including the 1,000 Genomes Project, ClinVar database or the Exome Aggregation Consortium (ExAC). These PLOD1 variants in the present cases are possibly causative for kEDS for the following reasons: First, in-frame deletions in non-repeat regions result in mutant protein sequences differed from the wild-type with the deletion of one or more amino acid residues, which are considered one of the evidences of pathogenicity. The novel p.Phe724del variant is located in a highly conserved region of oxoglutarate/iron-dependent dioxygenase, prolyl-4-hydroxylase, alpha subunit and the novel p.Leu309_Leu311del variant is in nucleotide-diphospho-sugar transferase domain. These variants can lead to deficiency in lysyl hydroxylation result in kEDS.19 Second, considering that kEDS is an inherited autosomal recessive trait, the identified compound heterozygous variants inherited from each parent could be regarded as the patient’s cause of the disorder.
In conclusion, this is the first reported case of siblings with kEDS who had PLOD1 variants in Korea. We identified two novel variants in the PLOD1 gene. kEDS with PLOD1 mutations should be considered in patients with hypotonia, progressive kyphoscoliosis, joint hypermobility and skin fragility. This will help reduce the incidence of unnecessary neuromuscular work-up and increase the early detection rate.
 XML Download
XML Download