

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Menkes disease (MD) is a very rare hereditary multisystemic disorder of copper metabolism with an X-linked recessive inheritance.123 MD is alternatively described as “kinky hair disease,” because of the characteristic sparse and steely gray or bright-colored hair of the patients. MD is caused by a defect in the ATP7A gene, which encodes a trans-membrane copper-transporting P-type ATPase, ATP7A.456 Defective copper transport in MD leads to malfunction of one or more copper-requiring enzymes (cuproenzymes), including lysyl oxidase, cytochrome c oxidase, tyrosinase, extracellular superoxide dismutase, and peptidylglycine α-amidating monooxgenase.7891011 The diverse clinical features of MD are attributable to the malfunction of one or more cuproenzymes.12
Patients with MD typically present with progressive neurodegeneration, some connective tissue abnormalities, and characteristic “kinky” hair. Various kinds of urological complications are also frequently observed in MD, including bladder diverticula, bladder outflow obstruction, vesicoureteral reflux, renal rupture, and cryptorchidism.13 These conditions predispose patients with MD to recurrent urinary tract infections and renal parenchyma damage. However, systematic studies on urological complications have not been reported extensively.
In this study, we analyzed the clinical features, image findings, and outcomes of urological complications in 14 Korean pediatric patients with MD.
METHODS
A total of 14 unrelated Korean pediatric patients diagnosed with MD at Seoul National University Children's Hospital, Seoul, Korea or Pusan National University Children's Hospital, Yangsan, Korea between 2005 and 2017 were included in this study. The phenotypes of the patients, with focus on accompanying urologic problems, were analyzed by retrospective review of the medical records. Mutational analysis of the ATP7A gene was carried out by direct sequencing and/or multiplex ligation-dependent probe amplification.14 Genomic DNA was isolated from peripheral blood leukocytes. The cases of seven patients (patients 1, 3–6, 9, and 12) have already been reported.14
RESULTS
Initial presentation of the patients
Among the 14 patients, 13 were boys and one (patient 14) was a girl. The median gestational period and birth weight were 38 weeks (ranges, 36+1–41 weeks) and 3.06 kg (ranges, 2.50–3.80 kg), respectively. None of the patients had any serious perinatal problems. All the patients presented with neurologic deficits, including developmental delay (n = 7), seizures (n = 6), and hypotonia (n = 3). The onset age was within 4 months after birth in 12 patients, while the remaining two patients, one without a pathogenic mutation and the other being the girl patient, presented with developmental delay at the age of 10 months and 13 months, respectively. The initial serum copper level was available for 11 patients, with a median value of 17.5 μg/dL (ranges, 7–66 μg/dL; normal, 65–140 μg/dL). The initial serum ceruloplasmin level (n = 10) was ≤ 10 mg/dL (normal, 15–40 mg/dL) in all patients except the girl patient, who had normal initial serum copper and ceruloplasmin levels (Table 1).
Table 1
Phenotypes and genotypes of the patients

Mutational analyses of the ATP7A gene
Mutational study of the ATP7A gene was performed for 13 patients, and it revealed 11 different pathogenic mutations in 12 patients, including 4 large deletions, 2 missense mutations, 2 nonsense mutations, 2 frame-shifting short deletions, and 1 abnormal splicing mutation. Seven mutations were novel. No mutation was detected in patient 6, and the mutational study was not done for patient 12. Family history of MD was negative in all patients except one (patient 7), who had an affected younger brother. Genetic testing was performed on the family members of the patients, and the mother of patients 7 was found to be heterozygous carrier of the mutation found in her son (Table 1).
Urological problems
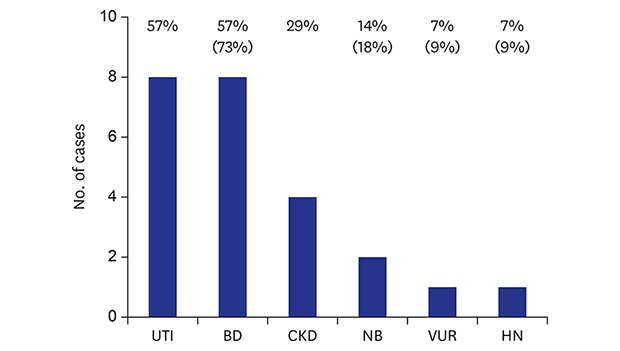
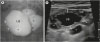
Imaging studies of the kidney and urinary tract were performed for 11 patients (patients 1–11), and bladder diverticula was the most common finding detected in eight patients (patients 1–8) (Fig. 1A). Neurogenic bladder dysfunction was noted in two patients (patients 6 and 7). In patient 6, ultrasonography revealed diffuse wall thickening of the urinary bladder (UB) with multiple bladder diverticula (Fig. 1B), and a urodynamic study revealed neurogenic overactivity of the detrusor muscle and synergic sphincter muscle without vesicoureteral reflux. Imaging studies of patient 7 revealed bilateral vesicoureteral reflux on voiding cystourethrography and diffuse wall thickening of the UB with bladder diverticulum on ultrasonography. However, an urodynamic study was not performed in patient 7 due to lack of parental consent. Bilateral hydroureter was detected in patient 3, and small renal cortical scar on the left upper pole of the kidney was detected on technetium-99m dimercaptosuccinic acid renal scan in patient 2. The imaging studies (n = 8) were performed chiefly as part of a diagnostic work-up for concomitant urinary tract infections. Urinary tract infections developed recurrently in three patients. Two patients received urological treatment: patient 6, who had severe neurogenic bladder dysfunction, underwent bladder diverticulectomy at the age of 5 years, and patient 7, who had bilateral vesicoureteral reflux and 19 episodes of urinary tract infection, underwent ureteroneocystostomy at the age of 1 year.
Fig. 1
Image findings of two common urological complications of Menkes disease, bladdr diverticuli and neurogenic bladder dysfunction. (A) Voiding cystourethrogram (patient 5) demonstrates multiple diverticuli (asterisks) arising from the UB. (B) Pelvic ultrasonogram (patient 2) with a high-frequency linear transducer reveals diffuse wall thickening (lines with a double arrow) of the UB and multiple diverticuli (asterisks).
UB = urinary bladder.
Other organ manifestations that were detected were pectus excavatum (n = 6), inguinal hernia (n = 5), intracranial hemorrhage/cephalhematoma (n = 4), hip dislocation (n = 2), and diaphragmatic hernia (n = 1) (Table 2).
Table 2
Associated abnormalities in the urinary tract and other organs

Disease course and prognosis
All patients had neurodevelopmental delay of various severity levels, and 11 patients took long-term antiepileptic drugs because of intractable seizures. Two patients (patients 8 and 14) were treated with periodic subcutaneous injections of copper-histidine, but the treatment did not affect the overall disease course. Five patients underwent tracheostomy and percutaneous endoscopic gastrostomy, and one other patient needed a nasal biphasic positive airway pressure support.
Two patients died at the ages of 10 and 11 years, respectively, at the end of palliative care at home, and six patients were lost to follow-up at a median age of 16 months (ranges, 8–38 months). The median current age of four surviving patients was 75 months (ranges, 5–83 months). The median estimated glomerular filtration rate (calculated by the revised Schwartz equation, n = 11) at the last follow-up was 118 mL/min/1.73 m2 (ranges, 72–327). Four patients had stage II chronic kidney disease (Table 2).
DISCUSSION
MD is a lethal multisystemic disorder of copper metabolism, presenting with progressive neurodegeneration and connective tissue disturbances, together with characteristic “kinky” hair, as the main manifestations.123 MD is inherited as an X-linked recessive trait and therefore the vast majority of patients are men. There was one girl patient (patient 14) in our study, who showed normal initial serum copper and ceruloplasmin levels and had milder symptoms and older age onset than the boy patients. The clinical symptoms of MD in girl are caused by skewed inactivation of the X chromosome that harbors the mutation15 or translocation between the X chromosome and an autosome.16 Patient 14 in our study had a missense mutation in ATP7A, without evidence of chromosomal translocation.
Patients usually present with early-onset neurodevelopmental delay and uncontrolled intractable seizures. The neurological deficits are all irreversible, and most of the severely affected patients died before the third year of life.3 Patients in this study also showed similar poor prognosis.
MD is caused by mutations in the ATP7A gene.1718192021 To date, 354 disease-causing or potentially disease-causing mutations in ATP7A have been reported (HGMD® Professional 2018.2, https://portal.biobase-international.com/hgmd/pro/start.php) in association with MD or its variant phenotypes: missense/nonsense mutations, 114 (32%); splicing substitutions, 69 (19%); gross deletions, 65 (18%); small deletions, 58 (16%); and others. The mutations are distributed almost equally throughout the gene, without any hot spot. In our study, large deletions were the most common of the mutations.
Urologic complications are frequent in MD, with bladder diverticula being the most common one.13 The ATP7A protein is an energy-dependent, transmembrane protein that is involved in the delivery of copper to the secreted copper enzymes and in the export of surplus copper from the cells. Defective cellular copper transport in MD leads to altered activity of various kinds of cuproenzymes and the consequent diverse phenotypes of MD.12 Lysyl oxidase, a copper-dependent and elastic-fiber-associated cuproenzyme, is known to be responsible for lysine-derived cross-linking of collagen and elastin in connective tissue. Defective function of lysyl oxidase in MD results in an altered elastic-fiber morphology, which leads to arterial tortuosities, bladder diverticula and other elastic tissue pathologies, including premature rupture of fetal membranes, cephalohematoma, subdural hematoma, abnormal facies, high-arched palate, emphysema, hernias, loose skin and joints, osteoporosis, petechial hemorrhage, poor wound healing, and central nervous system degeneration.122223 In 2006, Zaffanello et al.13 performed a retrospective review of 57 cases of MD, including 55 published case reports and two of their own cases, focusing on urological complications. They reported that the prevalence of bladder diverticulum was 38.6% of the total number of patients. However, most of the reports that they reviewed did not focus specifically on urological complication and therefore the prevalence of urological complications may have been underestimated. In addition, urological imaging studies are usually performed only in patients with urological problems, including urinary tract infections, voiding problems, etc. Therefore, urological complications are more likely to be found in older patients with longer follow-up periods. In fact, Zaffanello et al.13 reported that the number of urological complications increased progressively with the age of the patients. The prevalence of urological complications in our study was high: 8 (57.1%) of a total of 14 patients or 8 (72.7%) of 11 patients in whom urological evaluation was performed, which may be closer to the actual prevalence. A longitudinal cohort study, rather than a cross-sectional study, is needed to clarify this issue.
The diverticula in MD are usually multiple and vary in size and location, as shown in our study. Although the pathogenesis of bladder diverticula in MD has not been fully elucidated yet, it may result from impaired elasticity of the bladder muscle layer, caused by lysyl oxidase dysfunction,12 as well as disturbed innervation of the bladder in association with severe progressive neurodegenerative changes.24 In our study, two patients had neurogenic bladder dysfunction. Since lysyl oxidase activity cannot be corrected by parenteral copper administration, connective tissue laxity and the progression of urological complications cannot be completely improved.25 However, Zaffanello et al.13 showed that the progression of urological complications may be delayed with copper treatment. In our study, only two patients (patients 8 and 14) were treated with parenteral copper: patient 8 demonstrated bladder diverticula on imaging studies performed at the age of 17 months after an episode of urinary tract infection, and patient 14, a girl patient with a mild phenotype, did not undergo urological imaging studies because of the absence of urinary tract infection.
Bladder diverticula result in urinary stasis with a high residual urine volume, which leads to infections and further bladder dysfunction. Therefore, the main goal of management in patients with bladder diverticula is to accomplish complete bladder emptying with clean intermittent catheterization or open surgical drainage in severe cases.262728 Two of our patients underwent urological intervention. However, because of its rarity, no consensus on the treatment of bladder diverticula in patients with MD is available. Furthermore, whether surgical treatment is warranted given the propensity of bladder diverticula to recur and the generally short expected life span of these patients remains to be elucidated.29 Therefore, surgical intervention should be considered based on the clinical condition of each patient, including the severity of urological problems, expected life expectancy, and tolerance to general anaesthesia.2930
In conclusion, urological complications are very frequent in MD, with bladder diverticula being the most common. Bladder diverticula predispose patients with MD to urinary stasis, recurrent urinary tract infections, and renal parenchymal damage. Therefore, urological imaging studies and appropriate management of urological complications are required in all patients with MD.
 XML Download
XML Download