

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Renal cell carcinoma (RCC) affects more than 270,000 individuals annually worldwide, and is attributable for nearly 120,000 deaths each year.1 In Korea, age-standardized incidence rate and age-standardized mortality rate were 5.7 (men, 8.2; women, 3.4) and 1.0 (men, 1.7; women, 0.5) per 100,000 in 2015, respectively.2 Clear-cell RCC (ccRCC) is the most common type of RCC and accounts for 70% of cases.3 Although patients with small, localized (stage I–III) ccRCC treated surgically have excellent 5-year survival rates, around 30% of patients treated for localized disease will relapse.45 Similar to other tumor entities, ccRCC presents with significant clinical heterogeneity, which ranges from indolent to highly aggressive.6 The identification of accurate predictors of clinical outcome is imperative to determine individualized follow-up strategies and to facilitate counseling regarding adjuvant therapy. At present, prediction of tumor relapse for patients with surgically treated localized ccRCC is based on clinical and pathological features, including TNM stage, nuclear grade, which can be subject to inter-observer variability and might not entirely account for individual tumor biology.789 These limitations have led to much research aimed at identifying more accurate molecular markers that lead to better identification and counseling for patients with RCC. Our ability to better manage patients with RCC has been largely driven by a better understanding of the genetic and epigenetic modifications.101112 Epigenetic gene silencing is a molecular mechanism of silencing a gene by methylating its promoter region.13 Although previous epigenetic studies have identified a number of frequently methylated genes in RCC, few studies have determined the prognostic implications of DNA methylation profile in RCC.141516
The present study aims to identify clinical diagnostic, prognostic, and predictive methylation markers in surgically treated ccRCC. We performed comprehensive DNA methylation array analyses to detect differentially methylated genes in ccRCC and assessed the implications of novel candidate genes as prognostic methylation markers for surgically treated ccRCC.
METHODS
Patients and tissue samples
Human kidney specimens from 12 pairs of ccRCC and normal tissue (array set) and 152 ccRCC including 25 pairs of ccRCC and matched normal tissue (validation set) from primary, histologically-proven ccRCC who underwent radical or partial nephrectomy were collected between September 1997 and December 2014 at our institution. The pathology samples were independently re-examined by a pathologist who was unaware of the use of the clinical data and confirmed the presence of tumor or normal tissue. To reduce confounding factors affecting the analyses, patients had to fulfill the following criteria to be enrolled in this study: 1) clinically and pathologically T1 to T4 without LN or distant metastasis; 2) histologic pure clear cell RCC; 3) minimum follow-up period of 3 months; 4) simple tumor enucleation and positive surgical margin at final pathology were excluded to avoid biasing the survival estimates.
Surgical procedures performed included pure laparoscopic, hand-assisted laparoscopic, robot-assisted and open approaches. Pathological staging was performed based on the 7th edition of the American Joint Committee on Cancer classification system, and histological differentiation was graded according to the Fuhrman nuclear grading system.1718 After surgery, each patient was monitored according to the standard guidelines.1920 Distant metastasis was defined as lymph-node and/or other organ (lung, brain, contralateral kidney, etc.) metastasis, but local recurrence was not included in this definition.
DNA methylation profiling
Twenty-four matched DNA samples (12 ccRCC and 12 matched normal-surrounding kidney tissues) were used for DNA methylation profiling. Twelve ccRCC tissue samples were randomly allocated with pathologic stage. Genomic DNA (gDNA) was extracted by standard methods by using the Wizard Genomic DNA Purification System (Promega, Madison, WI, USA). Bisulfite-modified gDNA was prepared using EZ DNA Methylation-Lightning kit (Zymo Research, Orange, CA, USA) according to the manufacturer's instructions. The methylation status was assayed using the Infinium HumanMethylation450 BeadChip array (Infinium Methylation 450K; Illumina Inc., San Diego, CA, USA), which enables interrogation of the methylation status of more than 480,000 CpG sites distributed over the whole genome. Fluorescence signals corresponding to C- or T-nucleotides were measured, and the data were used to assign a quantitative measure of methylation level of specific CpG islands.
Selection of candidate methylation-silenced genes
The β value represents a quantitative measure of the DNA methylation level of specific CpG islands and ranges from 0 (completely unmethylated) to 1 (completely methylated). Candidate methylation-silenced genes were selected with next criteria: 1) a difference in DNA methylation levels between ccRCC and matched normal kidney (∆β value) > 0.25; 2) a mean β value for matched normal kidney < 0.15.
Pyrosequencing analysis
The DNA methylation status of candidate methylation-silenced genes was specifically assessed by pyrosequencing analysis of 152 human kidney specimens using a PyroMark Q96 ID instrument (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Each primer was designed using Pyrosequencing Assay Design Software v2.0 (Qiagen). PSQ primers were designed to encompass the CpG island loci assayed on the Illumina Infinium array. Primers were designed using NCBI Reference Sequences build version 37. PCR reactions were carried out in a volume of 20 µL with 20 ng or more converted gDNA, PCR premixture (Enzynomics, Daejeon, Korea), 1 µL of 10 pmole/µL forward primer, and 1 µL of 10 pmole/µL biotinylated reverse primer. The thermocycling parameters were as follows: denaturation at 95°C for 10 minutes; followed by 45 cycles of 95°C for 30 seconds, annealing at 59°C for 30 seconds, and 72°C for 30 seconds; and a final extension at 72°C for 5 minutes. The primer sequences are described in Supplementary Table 1.
Statistical analysis
The DNA methylation profile data were normalized using quantile normalization in the R language environment (version 2.10.0, available at http://www.r-project.org/).
The differences in continuous variables between groups were assessed using a two sample t-test or ANOVA. Receiver operating characteristic curves were constructed to estimate the capability of candidate markers for the prediction of metastasis, and to determine the optimal cut-off point for dividing patients into subgroups (hypomethylation or hypermethylation) with the highest combined sensitivity and specificity. To integrate the methylation status of the methylation markers with prognostic relevance, each patient's methylation score (M score) was calculated as the sum of the methylation levels of selected genes multiplied by the corresponding regression coefficients derived from the Cox regression analysis used to assess the predictive value of each gene for prediction of distant metastasis.21 The Kaplan-Meier curves were used to estimate time to metastasis according to methylation status, and differences were evaluated using log-rank tests. The prognostic value of methylation status was evaluated using the multivariate Cox proportional hazards regression analyses. Statistical analysis was performed using SPSS 21.0 software (IBM, Armonk, NY, USA). A P value < 0.05 was considered statistically significant.
Ethics statement
The study was carried out in agreement with applicable laws and regulations, good clinical practices, and ethical principles as described in the Declaration of Helsinki. The Institutional Review Board (IRB) of Chungbuk National University approved this protocol (IRB No. 2010-01-001), and written informed consent was obtained from each subject. Collection and analysis of all samples was approved by the IRB of Chungbuk National University.
RESULTS
Methylation profiling and identification of candidate silenced genes in primary ccRCC
The methylation profiles of 12 pairs of ccRCC and surrounding normal kidney were analyzed by the Infinium HumanMethylation450 array (Fig. 1). Using selection criteria (△ β value > 0.25 and mean β value of matched normal kidney < 0.15), 104 unique CpG island loci that were hypermethylated in ccRCC compared with the NCs were identified. Of these, pyrosequencing analyses of three out of top ranked 5 candidate genes, (Zinc finger protein 278 [ZNF278], Family with sequence similarity 155 member A [FAM155A] and Dipeptidyl peptidase 6 [DPP6]) were technically available (Table 1).
Fig. 1
Heat map for hierarchical clustering of human kidney specimens from 12 pairs of clear cell renal cell carcinoma and normal tissue.

Table 1
β-value differences between tumors and normal controls

| Genes | β-valuea | Differential | P value | |
|---|---|---|---|---|
| Normal control | ccRCC | |||
| ZFP28 | 0.0654 | 0.361 | 0.295 | < 0.001 |
| FAM155A | 0.132 | 0.418 | 0.286 | 0.022 |
| DPP6 | 0.133 | 0.388 | 0.255 | < 0.001 |
ccRCC = clear cell renal cell carcinoma, ZFP28 = Zinc finger protein 28, FAM155A = family with sequence similarity 155, member A, DPP6 = dipeptidyl peptidase 6.
aThe β value represents a quantitative measure of the DNA methylation level of specific CpG islands and ranges from 0 (completely unmethylated) to 1 (completely methylated).
Validation of methylation array findings in an independent primary ccRCC
To validate methylation array findings, we used the independent validation cohort of 25 pairs of ccRCC and matched normal tissue with PSQ (Table 2). The methylation level of ZNF278, FAM155A and DPP6 was higher in ccRCC than normal kidney in independent validation cohort (each P < 0.001) (data not shown).
Table 2
Clinicopathologic characteristics of array and validation cohort

Association between methylation levels and tumor aggressiveness
To evaluate the relationship between methylation status of candidate genes and clinicopathological factors, such as pathologic T stage and Fuhrman grade. Clinicopathologic characteristics of independent primary ccRCC are listed in Table 2. The hypermethylation of three genes were associated with advanced tumor stage and higher tumor grade (all P < 0.05) (Table 3).
Table 3
Relationship between ZNF278, FAM155A and DPP6 methylation and clinicopathological parameters in surgically treated clear cell renal cell carcinoma

Methylation status as a predictor of distant metastasis
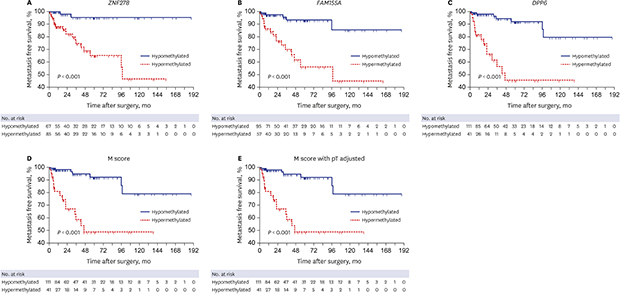
During median follow-up of 39.2 (interquartile range, 15.4–79.1) months, 22 (14.5%) patients experienced distant metastasis. Kaplan-Meier estimates identified significant differences in metastasis free survival according to methylation status of three candidate gene (log-rank test, all P < 0.001) (Fig. 2A-C). Integrate methylation score of three candidate genes, M score and adjusted-M score also showed identical results (log-rank test, all P < 0.001) (Fig. 2D and E). On multivariate analysis, hazard ratio (HR) was determined separately according to the methylation status of each gene and the clinicopathological factors. Multivariate analysis identified the methylation status of candidate three genes in each (ZNF278, HR, 7.012, P = 0.008; FAM155A, HR, 7.080, P = 0.008; DPP6, HR, 6.444, P = 0.011) (data not shown), or in a integrate methylation score of three candidate genes (M-score, HR, 3.804; P = 0.006) (Table 4) as an independent predictor of distant metastasis.
Fig. 2
Kaplan-Meier estimation for development of metastasis surgically treated clear cell renal cell carcinoma according to methylation status of three candidate gene. (A) ZNF278, (B) FAM155A, (C) DPP6, (D) M score and (E) M score with pathologic T stage adjusted.

Table 4
Multivariate Cox regression analysis for the prediction of metastasis in surgically treated clear cell renal cell carcinoma

HR = hazard ratio, CI = confidence interval, HTN = hypertension, DM = diabetes mellitus, BMI = body mass index, TNM = tumor node metastasis.
aM score was calculated with the sum of the levels of methylation levels of three candidate genes multiplied by the corresponding regression coefficient derived from the Cox regression analysis.
DISCUSSION
The present study aims to identify clinical diagnostic, prognostic, and predictive methylation markers in surgically treated ccRCC. We performed comprehensive DNA methylation array analyses and identified 3 novel methylated loci, ZNF278, FAM155A and DPP6. The validity of these candidate methylation markers was determined in a relatively large independent ccRCC. Notably, the methylation status of these candidate genes, either single or combined was closely related to not only tumor histology but also development of metastasis in surgically treated ccRCC. Our findings suggest the utility for the DNA methylation patterns in these genes as clinically useful surrogate markers, as well as new molecular pathways for further investigation as therapeutic targets in ccRCC.
Aberrant methylation of promoter CpG islands is an important inactivation mechanism of tumor suppressors and tumor-related genes.22 Methylation-induced silencing in early phases of tumorigenesis could potentially be used as markers for identifying individuals at increased risk of developing malignancy or for aiding in the diagnosis of early malignancy, whereas those genes undergoing methylation during the progression of malignancy could potentially be used as prognostic markers.2324 Genome-wide methylation profiling has been used as an approach for the identification of prognostic markers in different cancers evaluating thousands of CpG sites simultaneously.25 Although a number of hyper- or hypomethylated loci have been also identified for renal cell RCC, prognostic methylation markers of RCC that can provide useful information about survival and treatment options at diagnosis remains a major clinical challenge.1626
Candidate-gene approaches have disclosed several genes frequently methylated in ccRCC, including CDH1, APAF1, COL1A1, DKK2, DKK3, SFRPI, SFRP4, SFRP5, and WIF1.26 Genome-wide approaches have uncovered novel genes and pathways involved in ccRCC pathogenesis. McRonald and colleagues performed high-throughput epigenetic profiling using the Illumina Goldengate Methylation Array in 29 RCC from von Hippel-Lindau (VHL) disease and 20 sporadic ccRCC with wild type VHL and 13 sporadic papillary RCC to elucidate the differences in tumorigenesis mechanisms dependent on VHL gene status. They observed differing patterns of tumour-specific CpG methylation in VHL and non VHL ccRCC and papillary RCC.27 Morris et al.14 conducted the first whole-genome expression microarray study using methylated DNA immunoprecipitation (MeDIP) with whole-genome microarray analysis to identify differentially methylated regions in RCC. They identified a number of genes, including KLHL35, QPCT, SCUBE3, ZSCAN18, CCDC8, FBN2, ATP5G2, PCDH8 and CORO6 were frequently methylated in RCC and promoter hypermethylation of these genes resulted in significant reduction of their expression level. Recent study by Ricketts et al.15 have performed genome-wide methylation profiling of RCC using the HumanMethylation27 BeadChips in 38 sporadic RCC and 9 age-matched normal kidney controls. Eight novel ccRCC TSG candidates were identified, including OVOL1, DLEC1, BMP4, SST, TMPRSS2, TM6SF1, SLC34A2, and COL1A2, which demonstrated methylation in kidney cancer cell lines, re-expression in kidney cancer cell lines after 5-aza-20-deoxycytidine treatment, and tumor-specific methylation. Moreover, OVOL1 knockdown increased c-Myc mRNA levels which may lead to c-Myc pathway activation. We also performed the Illumina BeadArray technology to directly assay methylated CpG status, and identified 3 novel methylated loci, ZNF278, FAM155A, and DPP6. To the best of our knowledge, theses 3 loci have not previously been reported in RCC. From a prognostic point of view, the methylation status of these candidate genes, either single or combined was closely related to not only tumor histology but also development of metastasis in surgically treated ccRCC.
This study has several limitations and strengths. The β values of our three candidate genes are not high. We can't explain exactly the reasons, but this may be originated from the tumor characteristic of RCC or our relatively small number of methylation profiling. Second, there is currently only limited information available in the literature on the specific function of three candidate genes. ZNF278 also named POZ/BTB and AT-hook-containing zinc finger protein (PATZ), is a recently identified transcription factor with seven C2H2-type zinc fingers. Although the physiological role of ZNF278 is not clear, experimental evidence suggests that it is a potential transcription repressor. Previous study observed up-regulation of ZNF278 expression in human colorectal cancer tissues.28 The specific function of FAM155A has not been determined. DPP6 encodes protein dipeptidyl-peptidase 6, which binds to specific voltage-gated potassium channels and alters their expression and biophysical properties. A recent study on core signaling pathways in human pancreatic cancers suggested that DPP6 might play a crucial role in regulation of invasion of pancreatic cancer cells.29 Additional studies are needed to define the precise biological functions of these novel biomarkers on tumorigenesis and prognosis in ccRCC. Nonetheless, the present study represents an important step towards the clinical use of prognostic methylation markers in ccRCC.
In conclusion, the present study identified potential methylation markers which could represent the aggressive tumor phenotype and early development of distant metastasis in ccRCC. These potential methylation markers, either alone, or integrate methylation score, could offers a chance to identify cancer-specific diagnostic and prognostic tools, in addition to novel targets for development of individualized therapeutic and prevention regimens.
 XML Download
XML Download