

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a progressive and usually lethal lung disease characterized by fibroblast proliferation and extracellular matrix accumulation (1). Its pathogenesis remains poorly understood but is associated with a predominance of Th2 cytokine and a paucity of Th1 cytokine (2-4). Studies on immunopathology (2) and m-RNA expression (3, 4) have revealed that the lung tissues of IPF patients show abnormally high levels of interleukin (IL)-4 and IL-5, and a low level of interferon-γ (IFN-γ). Flow cytometry analysis of intracellular cytokines showed a similar pattern in the bronchoalveolar lavage (BAL) cells of IPF patients (5). The role of Th2 cytokines, especially IL-4 and IL-13, in pulmonary fibrosis has been clearly demonstrated by both in vivo and in vitro experiments (6, 7). These two cytokines function via complex receptor systems that include IL-4 receptor alpha (IL-4Rα), and IL-13 receptor alpha1 (IL-13Rα1) (8). IL-13Rα1 binds IL-13 in a complex with IL-4Rα1 (9, 10). IL-13 receptor α2 (IL-13Rα2) has a short cytoplasmic tail without intracellular signaling motifs; thus, it may function as a decoy receptor that inhibits the effects of IL-13 (10, 11). The down-regulatory effect of IL-13Rα2 has been demonstrated in animal models with IL-13-mediated tissue fibrosis (12) but has not been shown in humans.
IPF and nonspecific interstitial pneumonia (NSIP) are the two common types of idiopathic interstitial pneumonia (IIP). However, compared with IPF, NSIP leads to less fibrosis (13, 14) and shows a better response to anti-inflammatory agents such as steroids and anti-proliferative drugs (15, 16). Therefore, the two diseases may induce different patterns of fibrosis-related cytokines. It has already been demonstrated that NSIP and IPF lead to different levels of inflammatory cytokines and CXC chemokines, such as IL-6, IL-8, epithelial neutrophil-activating peptide 78, interferon gamma-inducible protein 10, RANTES, and monocyte chemotactic protein-1 (17-20), but little is known about how Th2 and Th1 cytokines and their receptors differ between the two conditions.
We hypothesized that IFN-γ, IL-4, and IL-13 may be regulated differently and that IL-13Rα1 and IL-13Rα2 may be expressed differently in IPF and NISP patients.
To test this hypothesis, we measured IL-4, IL-13, and IFN-γ levels in the BAL fluid of IPF and NSIP patients and normal controls, and compared IL-13Rα1 and IL-13Rα2 expression in diseased lung tissues using immunohistochemical staining.
MATERIALS AND METHODS
Study populations
The study population consisted of 16 patients with IPF, ten with idiopathic NSIP, and eight normal controls. All subjects were given a chest radiograph, HRCT, pulmonary function test, and blood gas analysis (Table 1). The diagnoses of IPF and NSIP were based on the international consensus statement (21). Two of the NSIP patients had inflammation and fibrotic type pneumonia (group II) and eight had interstitial fibrotic type pneumonia (group III). BAL and open -lung or video-guided thoracoscopic biopsies were performed on all IIP patients. BAL was performed at least 3 days before lung biopsy and without any immunosuppressive therapies. In normal subjects, BAL was performed on the right middle lobe. Exclusion criteria consisted of other known causes of interstitial lung disease, such as drug reactions, environmental exposures, and collagen vascular diseases. The normal controls were all healthy volunteers, free of symptoms, not taking any medications, and who had normal chest radiographs. For immunohistochemical staining, the healthy tissues of biopsies of solitary pulmonary nodules were used.
Measurements of IL-13, IL-4, and IFN-γ concentrations in Bronchoalveolar lavage (BAL) fluids
BAL was performed as previously described (17), using a flexible fiberoptic bronchoscope (Olympus B2-10; Olympus Optical Co., Tokyo, Japan). Briefly, all study subjects were pretreated with atropine (0.5 mg) immediately before the BAL procedure. Bronchial trees were instilled four times with 50 mL of normal saline, and fluid was removed each time by gentle suction with negative pressure below 50 mmHg. The total volume of recovered fluids was measured, and the supernatant was separated from the cell pellets by centrifugation at 500×g for 5 min. The supernatants were frozen and stored at -80℃ for later use. A differential count of 500 cells was performed on slides prepared by cytocentrifugation and Diff-Quik staining (Scientific Products, Gibbstowne, NJ, U.S.A.). No complications were experienced during the BAL procedures.
The IL-13, IL-4, and IFN-γ levels were measured using quantitative sandwich enzyme immunoassays with ELISA kits (BD Bioscience, San Diego, CA, U.S.A.), according to the manufacturer's recommendations. The detection limits were 2.0 pg/mL, 3.5 pg/mL, and 1.0 pg/mL for IL-4, IL-13, and IFN-γ, respectively, and values below these thresholds were assigned a value of 0 pg/mL. For validation of assay, we performed spiking experiment using 10 samples and the recovery rates of IL-4, IL-13, and IFN-γ were 86.2±0.1%, 93.5±0.6%, 89.3±0.8%, respectively. The inter-assay coefficient of variability for all assays was less than 10%, and the intraassay coefficient of variability was less than 10% across the range of concentrations.
Immunohistochemical staining
A VectaStatin goat ABC Elite kit (Vector Laboratories, Burlingame, CA, U.S.A.) was used for immunostaining. Tissue sections (5 µm) were deparaffinized, endogenous peroxidase was blocked with 1.4% H2O2 in MeOH for 30 min, and nonspecific binding was blocked with 1.5% normal rabbit serum for 30 min. The sections were incubated with goat polyclonal antihuman IL-13 antibody (1:100, Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and goat polyclonal antihuman IL-13Rα1 and IL-13Rα2 antibody (1:200, R&D, Minneapolis, MN, U.S.A.) at 4℃ for 16 hr. After washing with TBS, the sections were sequentially incubated with biotinylated rabbit anti-goat immunoglobulin G (H+L) (1:200, Vector Laboratories, Burlingame, CA, U.S.A.) and avidin-biotin peroxidase complex (1:50, ABC kit, Vector Laboratories) for 30 min. The color reaction was developed with 3,3'-diaminobenzidine tetrachloride (Zymed Laboratories, South San Francisco, CA, U.S.A.), and Harris hematoxylin stain was applied as a counter stain. The intensity of immunostaining was graded from absent (-) to strongly positive (+++) for the following tissues: alveolar epithelium, bronchial epithelium, smooth muscle, alveolar macrophage, endothelial cells, and interstitium. Grading was as follows: (-), no staining; (±), very faintly brown; (+), faintly brown; (++), moderately brown; and (+++), dark brown. Histological sections were assessed independently by two observers who were blind to the clinical information of the study subjects.
Statistics
The SPSS/PC+ program (Chicago, IL, U.S.A.) was used for statistical analyses. Differences between groups were compared with the nonparametric Kruskal-Wallis test for continuous data. Significant relationships between groups were further analyzed using the Mann-Whitney U test. Spearman's rank correlation was used to analyze the relationships of IL-4, IL-13, and IFN-γ concentrations to the lung functions. Differences were considered significant at P<0.05. The data are expressed as mean±SEM.
RESULTS
Clinical profiles of study subjects and cellular proportions in BAL fluids
The clinical and BAL profiles are summarized in Table 1. Age, smoking status, and sex ratio were comparable among the three groups. The NSIP and IPF patients had a significant reduction in forced vital capacity (FVC), total lung capacity (TLC), and diffusing capacity of carbon monoxide (DLCO) compared with controls (P<0.05). Both the cell number and percentage of neutrophils in 1 mL of BAL fluid were significantly higher in patients with NSIP and in those with IPF than in control subjects (P<0.01 and P<0.05, respectively). There were no difference in the percentages of lymphocytes and macrophages between NSIP and IPF patients, but IPF patients had a significantly higher percentage of eosinophils than controls (P<0.01).
IL-4, IL-13, and IFN-γ concentrations in BAL fluids
IL-13 activity was detected in the BAL fluid of all patients and in seven of the eight control subjects. The IL-13 levels were higher in IPF patients than in NSIP patients (249.0±83.1 vs. 137.7±37.9 pg/mL, P=0.032) and controls (249.0±83.1 vs. 49.0±13.1 pg/mL, P=0.012). The IL-13 levels were also higher in NSIP patients than in controls (P=0.041; Fig. 1A). IL-4 activity was detected in 12 of 16 IPF patients, eight of ten NSIP patients, and three of eight controls. The mean IL-4 level was significantly higher in the IPF and NSIP groups than in the control group (16.3±4.5 and 12.3±3.35 pg/mL vs. 4.67±2.28 pg/mL, P=0.008 and P=0.034, respectively). The IL-4 level was not significantly different between the IPF and NSIP groups (P>0.05; Fig. 1B). IFN-γ activity was detected in 14 of 16 IPF patients, nine of ten NSIP patients, and seven of eight controls. Both patient groups had low INF-γ levels compared with the control group (7.72±3.02 and 3.82±1.59 pg/mL vs. 11.32±3.64 pg/mL; Fig. 1C), but only the difference between NSIP patients and controls was significant (P=0.047). The IL-13 level was inversely correlated with DLCO (% pred) (n=26, r=-0.58, P=0.014) and FVC (% pred) (n=26, r=-0.47, P=0.043) in NSIP and IPF patients (Fig. 2). Neither the IL-4 nor INF-γ level correlated with the physiological parameters (data not shown).
Expression of IL-13 and IL-13Rα1/α2 detected by immunohistochemical staining
Immunohistochemical staining of IL-13, IL-13Rα1, and IL-13Rα2 was performed in the lung tissues of controls (n=4), IPF patients (n=6), and NSIP patients showing fibrosis (n=6). To avoid the smoking effect, never smokers were selected from the study subjects. Control subjects had undergone a lobectomy to remove localized solitary pulmonary nodules (two lung cancer and two tuberculoma). Both IL-13 and IL-13Rα1 were strongly expressed in the bronchial epithelial cells, alveolar macrophages, endothelial cells, and smooth muscle of all patients (Fig. 3), although semiquantitative evaluation showed that their intensities were much stronger in IPF patients than in NSIP patients (Table 2). IL-13Rα2 showed the same pattern of expression as IL-13Rα1 but weaker staining in both patient groups (Table 2, Fig. 3). Neither IL-13Rα1 nor IL-13Rα2 expression was detected in the four control lung tissues.
DISCUSSION
We demonstrated that IL-4 and IL-13 levels were higher in the BAL fluids of NSIP and IPF patients than in that of healthy subjects and that INF-γ levels were lowest in the NSIP groups. These findings suggest that there is a predominance of Th2 over Th1 cytokines in the lungs of NSIP patients, as demonstrated in IPF patients of previous studies (2-4). In the present study, the mean IL-13 level was higher in patients than in controls, consistent with the reports of Jakubzick et al. (4), who demonstrated that IL-13 gene expression was higher in lung biopsies of IPF and NSIP patients. However, in our study, the mean IL-13 level was higher in IPF patients than in NSIP patients, whereas Jakubzick et al. (24) reported no significant differences in IL-13 gene expression between the two patient groups. In both studies, IL-4 levels were comparable between the two groups.
In addition, the IL-13 (and not IL-4) level was inversely correlated with physiological derangement, including decreases in DLCO and lung volume. This novel finding (Fig. 2) suggests that IL-13, rather than IL-4, may be associated with functional derangement in the patients with IPF and NSIP.
Several studies on animal models have suggested a comprehensive role for IL-13, rather than IL-4, in pulmonary fibrosis. Neutralization of IL-13, but not IL-4 alone, attenuated pulmonary fibrosis in a bleomycin-induced pulmonary fibrosis model (22). In response to fluorescein isothiocyanate exposure, IL-4 (-/-) mice developed fibrosis, whereas IL-13 (-/-) mice were significantly resistant to fibrosis (23). These results, and the fact that IPF is associated with more fibrotic lung lesion than is NSIP, suggest that IL-13, rather than IL-4, may be responsible for the degree of fibrosis in IIP patients, as seen in our study. Indeed, administrating a chimeric protein, comprising human IL-13 and a truncated version of an exotoxin from Pseudomonas (IL13-PE), as a therapeutic agent significantly attenuated fibrosis (24). IL-13 may induce pulmonary fibrosis directly (25) or indirectly through TGF (beta)-1 (26) and several types of MMP (27).
Receptor binding chains for IL-13 include IL-13 Rα1 and IL-13Rα2 (11). IL-13 Rα1 binds IL-13 with low affinity, whereas the co-expression of IL-13Rα1 and IL-4Rα results in the formation of a high-affinity receptor signaling complex that renders a variety of cells (e.g., mast, eosinophil, endothelial, fibroblast, smooth muscle, and epithelial cells) responsive to IL-13 (28). In the present study, both IL-13Rα1 and IL-13Rα2 were expressed in the bronchial epithelial cells, alveolar macrophages, endothelial cells, and smooth muscles and interstitium of all IPF and NSIP patients, consistent with Jakubzick et al. (4), who found that both proteins were strongly expressed in the interstitium of the lung and that IL-13Rα2 was also strongly expressed in the lung epithelium and in mononuclear cells.
The effects of IL-13 are mediated by a complex receptor system, which includes IL-4Rα and IL-13Rα1, via STAT6 activation (10, 11). IL-13Rα2 has a short cytoplasmic tail without intracellular signaling motifs that bind to STAT6 (10, 11) thus, this receptor was expected to compete with the IL-4Rα-/IL-13Rα1 complex and dampen the IL-13-mediated response (29). At present, however, it is not clear why the greatly increased expression of IL-13Rα2, particularly in IPF patients, fails to curtail the profibrotic actions of IL-13. It is possible that the natural antagonism of IL-13Rα2 is lost in patients with IIP as a result of post-translational modifications of this decoy receptor. Or perhaps, as a result of these modifications, the IL-13Rα2 subunit takes on an abnormal signaling role in IPF. Fichtner-Feigl et al. (30) recently demonstrated that IL-13 is involved in the activation of the TGFβ1 promoter via IL-13Rα2. Furthermore, blocking IL-13 Rα2 signaling with siRNA led to a down regulation of TGFβ1 production and collagen deposition in bleomycin-induced lung fibrosis (30). This result indicates that IL-13Rα2 can augment fibrosis via TGFβ modulation, which contradicts other studies (12, 29).
The present study did not directly evaluate the role of IL-13Rα2 as a protector against or inducer of fibrosis in IIP. We demonstrated the increased expression of IL-13Rα1 and IL-13Rα2 in the two types of IIP, and no expression in controls, indicating that both types of receptors are induced in the tissues of people with IIP. However, the intensity of these two receptors was significantly lower in NSIP patients than in IPF patients. Furthermore, IL-13Rα1 was strongly expressed in the epithelium, smooth muscle, and alveolar macrophages of both patient groups, whereas IL-13Rα2 was only weakly expressed (Fig. 2, Table 2), suggesting that differences in the expression of IL-13Rα1 and IL-13Rα2 may help explain the varying degrees of lung fibrosis between the two conditions. A possible limitation of our study is that the differences in IL-13 values for the BAL fluids of IIP patients and controls (as determined by ELISA) may have been affected by the presence of soluble isoforms of the IL-13 receptor subunits. There are many potential complications for measuring cytokine proteins in whole lung homogenates; for example, the presence of soluble decoy receptors and post-translational modifications of the cytokine proteins can render the proteins undetectable by ELISA. Small numbers of NSIP and IPF in this study also regarded another limitation.
In conclusion, IL-13 levels were significantly elevated and inversely correlated with DLCO and FVC in both IPF and NSIP patients. Moreover, IL-13Rα1 and IL-13Rα2 were strongly expressed in the lung tissues of IPF and NSIP patients. However, IL-13 and the two receptors were more strongly expressed in patients with IPF than in those with NSIP. These results suggest that the elevation of IL-13 elevation and the concomitant expression of IL-13Rα1 and IL-13Rα2 may be associated with the pathogenesis of both IPF and NSIP.

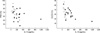



 XML Download
XML Download