

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Dear Editor,
CHARGE syndrome (CS; OMIM 214800) is a rare genetic disease encompassing multiple developmental anomalies, with an estimated incidence of 1:8,500–12,000 live births [1]. Diagnosis of CS relies on combinations of well-defined clinical features grouped into major and minor criteria (Table 1) [2]. The absence of one functional allele of CHD7 is responsible for 60–70% of CS cases [3]. CS cases are very rarely linked to alterations in other genes, such as EFTUD2 (OMIM 603892) [4]. CHD7 is one of the largest human genes, with a size of 188 kb, encompassing 38 exons. As a large-scale mutation scanning strategy, targeted gene panel sequencing has become the most straightforward approach.
In Indonesia, only one case of clinically diagnosed CS has been reported to date, without further investigation [5]. We provide a retrospective report of the molecular diagnosis of this patient based on next-generation sequencing (NGS). To our knowledge, this is the first report on the molecular diagnosis of CS in an Indonesian patient. This study was approved by the Ethics Board of the Faculty of Medicine, Swadaya Gunung Jati University, Indonesia (No. 64/EC/FK/IX/2017). Informed consent was obtained from the patient's parents.
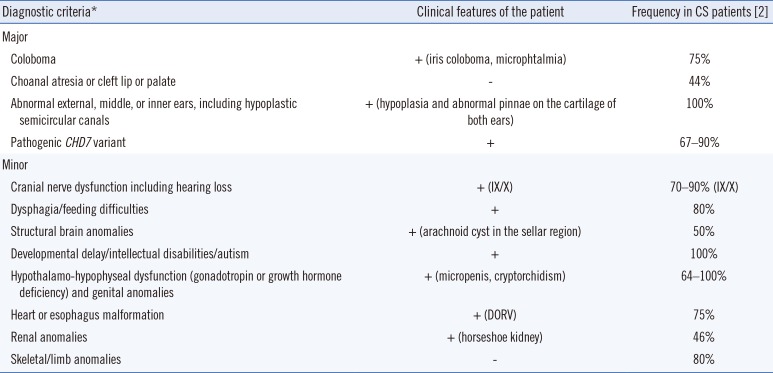
A five-year-old boy with multiple deformities was born in 2014 from non-consanguineous Indonesian healthy parents. The pregnancy was without complications, and delivery was spontaneous. Since the first week of life, the infant had feeding problems, short stature, and failure to thrive. The clinical diagnosis of CS was established in the first year by pediatricians at Diponegoro National Hospital, Semarang, Indonesia. He had two healthy brothers, and there was no family history of similar complaints, nor any other congenital abnormality in the three-generation family pedigree. The patient exhibited two major and seven minor clinical criteria (Table 1), confirming the diagnosis of CS.
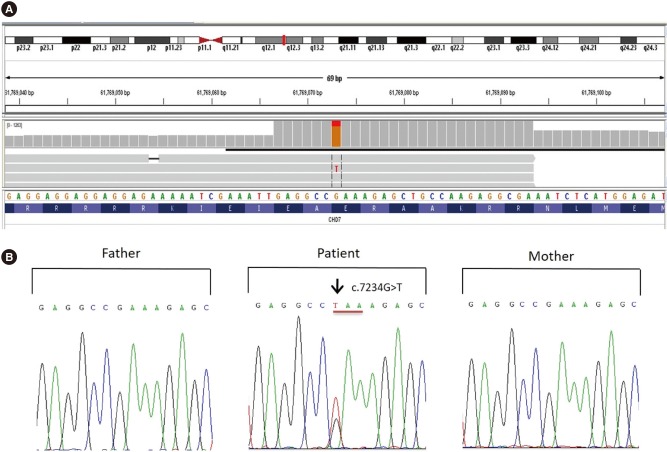
The patient had a normal male 46, XY karyotype (not shown). We determined the whole exonic and flanking intronic sequences of two genes involved in CS, CHD7, and EFTUD2. HOXA1 (OMIM 142955) was included in the panel as alterations in this gene are involved in features overlapping with CS [6]. For NGS, we used a panel containing 209 primer pairs in two pools (Ion AmpliSeq, Thermo Fisher Scientific, MA, USA). Sequencing was conducted on a Personal Genome Machine (PGM) sequencer (Ion Torrent, Thermo Fisher Scientific), according to the manufacturer's instructions. We evaluated the variants using the Alamut Visual 2.11 software (Interactive Biosoftware, Rouen, France). We identified a heterozygous nonsense variant in exon 34 of CHD7: NM_017780.3:c.7234G>T (p.Glu2412Ter; Fig. 1A). We confirmed this finding by Sanger sequencing of exon 34, using PCR primers CHD7-F 5′-GCCAGCCCATATAGCAGTAC-3′ and CHD7-R 5′-AACACAGCCCAGCATCGTGA-3′. This variant was confirmed as de novo as we did not detect it in the DNA of either of his parents (Fig. 1B). As this novel variant has not been reported before, we deposited it in the CHD7 database (https://molgenis51.gcc.rug.nl/, last accessed: October 02, 2018).
According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Standards and Guidelines for the Interpretation of Sequence Variants [7], this variant is classified as pathogenic: PVS1 (very strong evidence of pathogenicity because it is a null variant), PS2 (de novo, both paternity and maternity confirmed), PM2 (variant absent in control population), PP3 (deleterious effect confirmed by multiple computational evidence). Thus, we identified the genetic cause of CS in this patient, and we added this disease-causing CHD7 variant as a third major criterion for CS diagnosis.
The birth of a child with a severe genetic disease poses considerable psychological, social, and economic challenges to the family. A genetic counselor should provide recurrence-risk estimates to the parents in such cases. Even if the parents have had children affected by apparently de-novo
CHD7 alterations, there is still a recurrence risk of 1–2% because of somatic and germline mosaicism [8]. Therefore, noninvasive prenatal testing should be recommended to the mother during subsequent pregnancies to determine whether the fetus has a chance of having CS.
There are several approaches to establish a molecular diagnosis of clinically suspected CS. Sanger sequencing of 38 exons and flanking intronic sequences of CHD7 gene is considered obsolete, as it is more time-consuming and eventually more expensive than NGS [9]. Currently, sequencing of large panels of genes involved in intellectual disability is widely used, occasionally leading to the identification CS-causing CHD7 mutations [10]. However, as the clinical diagnosis of CS relies on well-established criteria, sequencing of CS-targeted gene panels appears more straightforward. Moreover, the lower-capacity NGS equipment allows testing at a lower cost than sequencing of large gene panels or genome-wide analysis. This approach could be applied in low- and middle-income countries as a cost-effective strategy for routine diagnosis of CS.
 XML Download
XML Download