

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired pluripotent hematopoietic stem cell (HSC) disorder associated with partial or absolute glycosyl-phosphatidyl inositol-anchored protein (GPI-AP) deficiency [12]. In PNH, a somatic defect in HSCs, which synthesize glycosyl-phosphatidyl inositol (GPI), results in defective GPI linkage of decay-accelerating factor (DAF) and membrane inhibitor of reactive lysis (MIRL) in red blood cells (RBCs). The lack of cell surface expression of these proteins leads to cell lysis by the complement system and destruction of large numbers of RBCs, resulting in hemoglobinuria [345]. The most common cause of PNH is somatic mutations associated with the X-chromosomal gene PIGA, which encodes one of the several enzymes required for GPI anchor biosynthesis [67]. Somatic PIGA mutations in PNH patients are manifold, the majority including insertions or deletions involving a single base, such as single base substitution, or several bases.
The clinical manifestation of PNH involves clonal expansion of GPI-AP blood cells, which is controlled by intrinsic and extrinsic factors that affect cell growth and survival [8]. However, little is known about the clonal expansion of GPI-AP deficient HSCs in relation to PIGA mutations and blood cell lineage. We investigated PNH clonal proliferation in the three cell lineages, RBCs, granulocytes, and T lymphocytes, by analyzing PIGA gene mutations and T-cell receptor (TCR) clonality between PNH/aplastic anemia (AA) and classic PNH.
METHODS
Patients
Peripheral blood samples were collected for routine flow cytometry screening for PNH from 24 patients at Seoul St. Mary's Hospital, Seoul, Korea, between January 2010 and April 2012. Samples remaining after the screening test were aliquoted at 200 µL/tube. Two or three microtubes were stored immediately at −20℃ until molecular analysis. Diagnosis was confirmed and assigned according to the guidelines for the diagnosis and management of PNH [9]. At sampling, no patient was actively infected, and no evidence for hereditary bone marrow (BM) failure syndromes was found. All patients provided written informed consent for clinical and molecular analyses. The study protocol was approved by the Institutional Review Board of The Catholic University of Korea, Seoul, Korea (KC12RISE0422).
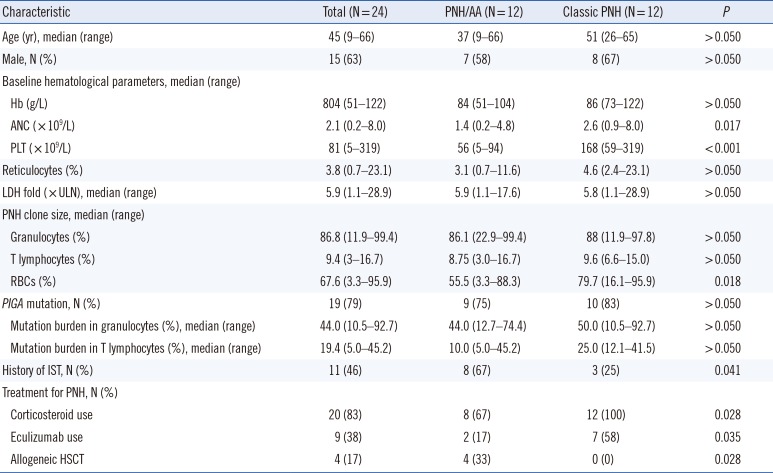
The patients were classified into two groups based on the presence of cytopenia at diagnosis of PNH: PNH with concomitant AA (PNH/AA, N=12) and classic PNH (N=12). Patients who met at least two of the three peripheral blood cytopenias (Hb level: ≤100 g/L, absolute neutrophil count [ANC]: 0.5–1.5×109/L, and platelet count [PLT]: 20–100×109/L) were classified as PNH/AA [10]. Patients with clinical evidence of intravascular hemolysis without any evidence of other BM failure were classified as classic PNH. There were significant differences between the groups in ANC and PLT. The number of patients who underwent previous immunosuppressive therapy and/or corticosteroid treatment was significantly higher in the PNH/AA than in the classic PNH group. The percentage of patients who were treated with eculizumab was higher in the classic PNH group, whereas allogeneic hematopoietic stem cell transplantation (HSCT) was performed exclusively in the PNA/AA group (Table 1).
Analysis of GPI-AP deficient cells
GPI-AP deficient granulocytes, T lymphocytes, and RBCs were analyzed by flow cytometry using specific monoclonal antibody cocktails: fluorescent aerolysin reagent (FLAER)-Ax488/CD24-PE for granulocytes, FLAER-Ax488/CD3-PE for T lymphocytes, and CD55-FITC/CD59-PE for RBCs. The FLAER-based flow-cytometric assay is recommended to screen for GPI-AP deficient white blood cells (WBCs) because it has higher sensitivity than that based on other GPI-APs, including CD55, CD59, CD24, and CD14, and can detect small clone sizes in PNH as well as AA or myelodysplastic syndrome [1112].
To analyze GPI-AP deficient granulocytes and T lymphocytes, 5 µL of appropriate monoclonal antibody was added to 0.5×106 WBCs and incubated for 20 minutes at 24℃. After RBCs were lysed using 2 mL of BD fluorescence activated cell sorter (FACS) Lysing Solution (BD Pharmingen, Franklin Lakes, NJ, USA), the cells were washed and analyzed on a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA, USA), according to the manufacturer's recommendation. For each tube, a minimum of 50,000 events for granulocytes or T lymphocytes was routinely collected. To analyze GPI-AP deficient RBCs, 20 µL of whole blood was diluted with 3 mL of phosphate-buffered saline (PBS) followed by the addition of 50 µL to one tube, and 5 µL of CD59-FITC and CD55-PE to the second tube. The samples were incubated in the dark for 60 minutes at 24℃ and washed twice with PBS by centrifugation as required to optimize separation of Type I, II, and III GPI-AP deficient RBCs. After washing, the RBC pellet was re-suspended in 0.2 mL of PBS, and 50,000 RBCs were acquired in the list mode.
Granulocytes and lymphocytes were initially selected by forward scatter and side scatter. Each cell lineage was assessed for combined expression of FLAER/CD24 (granulocytes) and FLAER/CD3 (T lymphocytes). The limit of detection (LOD) using FLAER as the most discriminant marker combined with CD24 or CD3 was determined as 0.1%. However, because FLAER affinity seems restricted to certain types of GPI anchors, making it unsuitable for evaluation of RBCs [13], we used both CD55 and CD59 for expression analysis for GPI-AP-deficient RBCs with an LOD of 3%.
Isolation of GPI-AP deficient granulocytes and T lymphocytes for molecular analysis
Granulocytes were isolated from EDTA-blood samples via standard density gradient separation using a mixture of sodium metrizoate and dextran 500 [14]. T lymphocytes were isolated from EDTA-blood samples using the EasySep Human T Cell Enrichment Kit (STEMCELL Technologies, Vancouver, Canada), according to the manufacturer's protocol. GPI-AP deficient granulocytes and T lymphocytes were additionally sorted into the FLAER(−)/CD24 and FLAER(−)/CD24 CD3(+) cell population, respectively, by flow cytometry using BD FACSAria II (BD Biosciences). Genomic DNA was extracted from the sorted GPI-AP deficient granulocytes and T lymphocytes using the QIAamp DNA Blood Mini Kit (Qiagen GmbH, Hilden, Germany).
PIGA mutation analysis
Genomic DNA isolated from granulocytes and T lymphocytes was used as a template for amplifying individual exons of PIGA, including entire exons and the flanking intron sequences, as described previously [15]. PCR amplicons were bi-directionally sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on an ABI PRISM 3130XL Genetic Analyzer (Applied Biosystems). The sequence electropherogram was analyzed using Sequencher Software 4.9 (Gene Codes, Ann Arbor, MI, USA). RefSeq ID: NM_002641.3 for PIGA was used for cDNA nucleotide numbering. Mutations were confirmed by sequencing via two or more independent PCRs. Genetic variations were compared with the public sequence databases including a database of single nucleotide polymorphisms (dbSNP, https://www.ncbi.nlm.nih.gov/snp/), Exome Aggregation Consortium (ExAc, http://exac.broadinstitute.org), 1000 Genomes Project Phase 3 database (1000 Genomes, http://phase3browser.1000genomes.org/index.html), and Korean Reference Genome Database (KRGDB, http://152.99.75.168/KRGDB/). Pathogenic mutations were predicted using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2) [16] and PROVEAN (http://provean.jcvi.org) [17]. We used numerical peak height data of the Sanger sequencing electropherogram to measure semiquantitative PIGA mutation burden [18].
TCR gene rearrangement assay
The TCR gene rearrangement assay was used to evaluate the clonality of GPI-AP deficient T lymphocytes. PCR was conducted on genomic DNA isolated from sorted GPI-AP deficient T lymphocytes using IdentiClone TCRB, TCRG, and TCRD Gene Clonality Assay (Invivoscribe Technologies, San Diego, CA, USA). The full set of reactions for TCR gene arrangements included one targeting TCRD (Vδ+Dδ+Jδ), one targeting TCRG 2.0 (Vγ+Jγ), and three targeting TCRB (TCRB Tube A: Vβ+Jβ1/2; TCRB Tube B: Vβ+Jβ2; TCRB Tube C: Dβ+Jβ1/2). The PCR products were analyzed on an ABI 3130XL Genetic Analyzer with GeneMapper software (Applied Biosystems). Criteria for defining a positive peak in TCRD and TCRB were as follows: (1) a single prominent positive peak within the valid size range for individual primer pairs and (2) products within the valid size range and with peaks of at least three times the amplitude of the third largest peak in the polyclonal background. The TCRG interpretation algorithm provided by the manufacturer was implemented in the worksheet to define peaks as positive for clonality in TCRG 2.0: (1) the D(x) value of the suspected clonal peak, as calculated within the locked portion of the worksheet, is ≥0.0419; (2) the relative peak ratio of the suspected clonal peak, compared with the smallest adjacent peak, is ≥4; (3) the relative fluorescence unit (RFU) of the suspected clonal peak is ≥20% of the RFU of the highest peak in that sample. All assays were conducted in duplicate.
Statistical analysis
As the laboratory findings were not normally distributed as indicated by Kolmogorov-Smirnov and Shapiro-Wilk tests, continuous variables were summarized as median with range and were compared using the Mann–Whitney U test. Pearson's chi-square test was used to compare categorical variables. Spearman's correlation was determined to estimate the strength and direction of an association that existed between two continuous variables, and a line of best fit was drawn through the data of two variables in a scatterplot. All tests were two-tailed, and P<0.05 was considered significant. All statistical analyses were conducted using MedCalc 12.7.2 (MedCalc Software, Ostend, Belgium).
RESULTS
GPI-AP deficient cell fraction in blood cell lineages
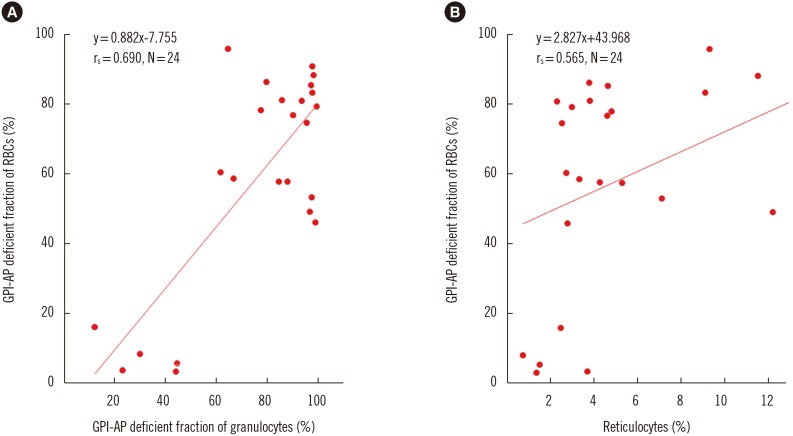
We excluded the patients treated with eculizumab from PNH clone size analysis according to cell lineage because eculizumab promotes the survival of GPI-AP-deficient RBCs compared with that of WBCs or lymphocytes [19]. The GPI-AP deficient cell fraction varied according to patient and cell types. The median GPI-AP deficient cell fraction was the highest in granulocytes followed by RBCs and T lymphocytes. GPI-AP-deficient RBCs were more numerous in classic PNH than in PNH/AA patients (P=0.018). The GPI-AP deficient granulocyte fraction correlated strongly with the clonal size of RBCs (rs=0.69, P=0.015). The GPI-AP deficient RBC fraction correlated moderately with percentage of reticulocyte (rs=0.565, P=0.004) (Fig. 1).
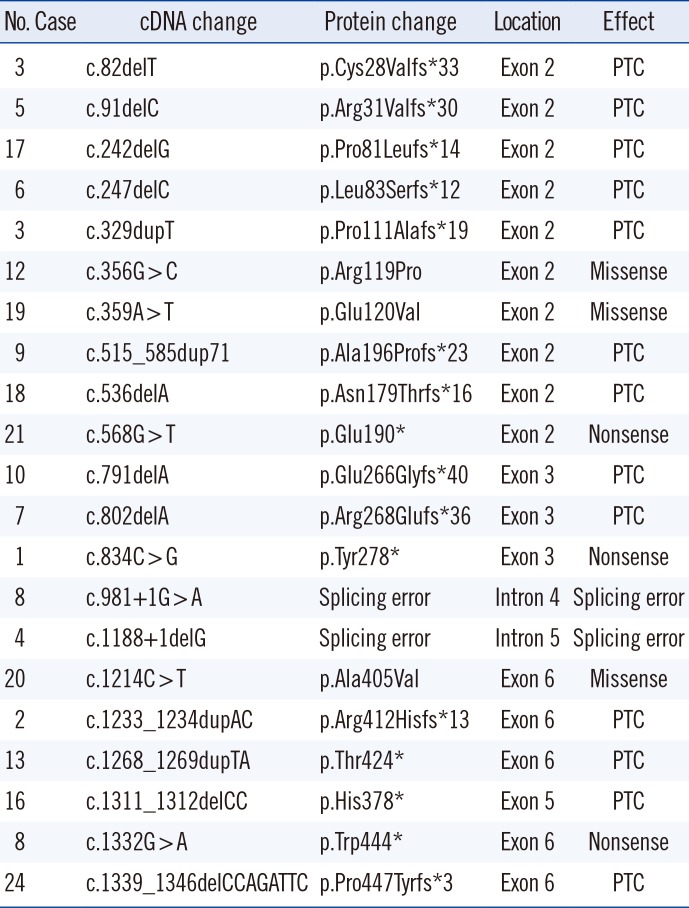
PIGA mutation profiles and mutation burden in blood cell lineages
Nineteen (79.2%) of the 24 PNH patients carried PIGA mutations. Among them, 17 patients carried a single heterozygous mutation, and two patients harbored two heterozygous mutations (Table 2). Each PIGA mutation was exclusive and commonly occurred in exon 2 (48%, 10/21). The frequencies of deletion, duplication, and base substitution in the PIGA gene were 43% (9/21), 19% (4/21), and 38% (8/21), respectively. These mutations led to deleterious effects, including premature terminations (N=13), nonsense mutations (N=3), splicing errors (N=2), and missense mutations (N=3). Among the three missense mutations, p.Arg119Pro and p.Glu120Val were deemed “probably damaging” by PolyPhen-2, with scores of 0.999 and 1.000, respectively, and were expected to be “deleterious” by PROVEAN, with scores of −6.137 and −6.530, respectively. In contrast, p.Ala405Val was considered “benign” by PolyPhen-2 (score 0) and “neutral” by PROVEAN (−0.871). The frequency of these three variants was searched in public sequence databases. p.Ala405Val was registered as a “likely benign allele” (RS201361742) in dbSNP, and its variant frequency was reported as 0.0003/27 in ExAc and 0.0003/1 in 1000 Genomes, but it was not registered in KRGDB.
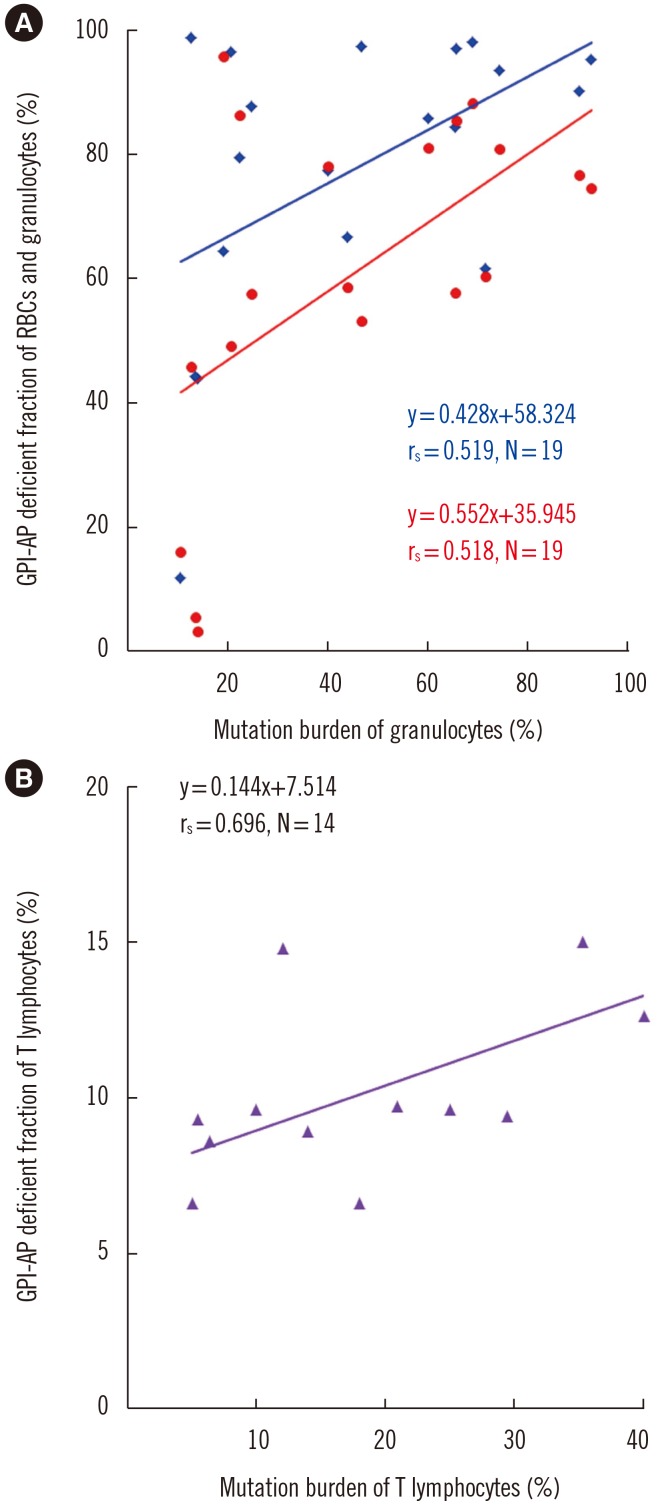
The incidence of PIGA mutation did not differ between PNH/AA and classic PNH. There were no significant differences between patients with and without PIGA mutation in blood cell counts, reticulocytes, and GPI-AP deficient cell fractions. Median PIGA mutation burden was significantly higher in granulocytes (44%) than in T lymphocytes (19.4%) (P=0.004). Interestingly, PIGA mutation burden in granulocytes correlated with the GPI-AP deficient fractions of RBCs (P=0.023) and granulocytes (P=0.049). Similarly, the mutation burden of T lymphocytes correlated with the GPI-AP deficient T lymphocyte fraction (P=0.006; Fig. 2).
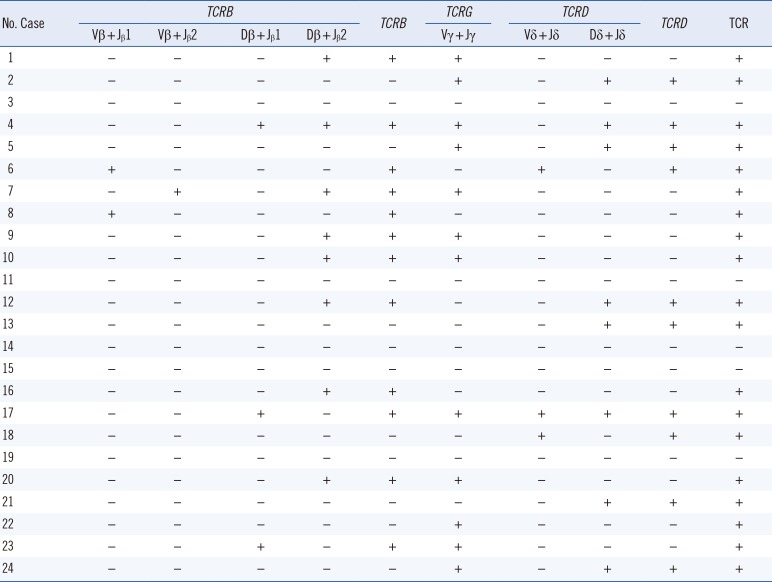
Association of TCR clonality and PIGA mutation
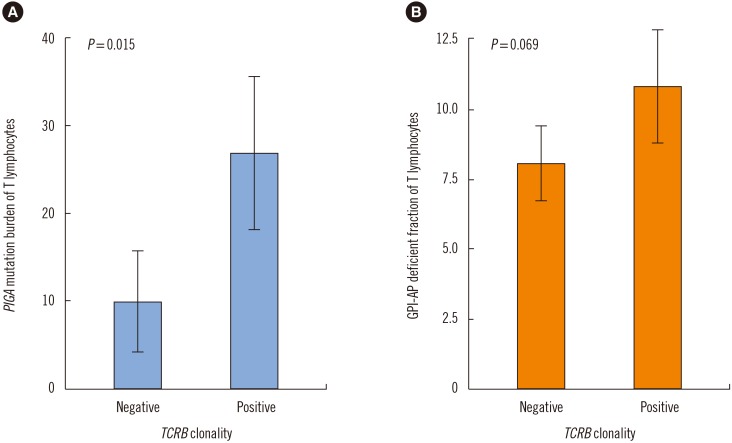
TCR clonality was detected in 19 (79.2%) out of the 24 PNH patients. TCRB, TCRG, and TCRD clonality was detected in 50%, 50%, and 41.7% of the patients, respectively (Table 3). Interestingly, PIGA mutation burden of T lymphocyte was more frequently detected in patients with clonal rearrangements in any of the TCR genes, compared with those with no TCR rearrangements (P=0.015). The PIGA mutation burden of T lymphocytes was higher in patients with clonal TCRB rearrangement than those with no TCR rearrangements (27.2% vs 8.2%; P=0.048; Fig. 3).
DISCUSSION
Individual somatic mutations throughout the coding region of the PIGA gene were identified in approximately 80% of our PNH patients. The majority (approximately 48%) of PIGA mutations were found on exon 2, which contains nearly a half of the coding region, although mutational hotspots were not observed. Like Nafa et al. [20], we frequently found small deletions or duplications/insertions in PIGA, which resulted in a frameshift in the coding region and a shortened, nonfunctional protein, except for the three missense mutations (two in exon 2 and one in exon 6). Although PIGA function is usually abrogated by these mutations, decreased PIGA activity due to missense mutations also induces PNH [21]. Because somatic PIGA mutations occur in only one or few HSCs, clonal expansion of these HSCs is necessary to develop PNH.
The mechanisms of clonal expansion have been studied actively to elucidate the pathogenesis of PNH [6722]. The consensus is that a PIGA mutation per se is not sufficient to induce clonal expansion; additional clonal selection by extrinsic factors and intrinsic clonal evolution is required [8]. More than 20 genes localized on various autosomes are known to mediate GPI biosynthesis and attachment to proteins, and mutation in those genes also triggers PNH development [23242526].
Consistent with previous studies, we revealed that PIGA mutations originated in a multipotent HSC; consequently, we could detect GPI-AP deficient fractions in all three lineages, including T lymphocytes, by flow cytometry as well as genetic analysis [272829]. The GPI-AP deficient fractions of blood cell lineages correlated with the PIGA mutation burden of the respective cell lineages. The GPI-AP deficient fraction was the highest in granulocytes, followed by RBCs and T lymphocytes. PNH was induced by GPI-AP-deficient RBCs chronically undergoing intravascular hemolysis with a lifespan reduced to 10% compared with that of normal RBCs [30]. By contrast, no evidence of reduced granulocyte lifespan was observed in PNH patients [31]. The GPI-AP deficient RBC fraction correlated moderately with percentage of reticulocyte, a representative marker of RBC proliferation, which suggested that clonal PNH RBCs show higher proliferation than normal RBCs. Patients with classic PNH had a larger GPI-AP-deficient RBC fraction than PNH/AA patients, which demonstrated that clonal PNH RBCs in classic PNH have higher proliferation activity.
Although both the GPI-AP deficient fraction and the PIGA mutation burden of T lymphocytes were lower than those of granulocytes, the TCR gene rearrangement assay demonstrated the clone formation capacity of GPI-AP deficient T lymphocytes. A previous study demonstrated that GPI-AP deficient T lymphocytes readily expanded and grew significantly in a T-lymphocyte enrichment culture system [29]. In addition, the correlation between PIGA mutation burden and TCR gene rearrangement suggested that PIGA mutant T lymphocytes themselves could produce a detectable clonal cell population. The fact that GPI-AP deficient and PIGA mutant T lymphocytes were only a minor fraction of lymphocytes in most patients may be explained by the long lifespan of lymphocytes [32] and the relatively lower rate of lymphopoiesis compared with myelopoiesis or erythropoiesis [33].
Potential limitations of this study include no identification of PIGA mutations in five patients with definitive GPI-AP deficient cells and no detection of any phenotypic differences between patients with and without PIGA mutation, including the development of PNH/AA. Therefore, we could not fully explain the clonal expansion of PNH cells based on PIGA mutation alone. Additional factors should be studied.
In conclusion, we revealed the presence of PIGA mutations in approximately 80% of PNH patients. The mutations were observed in both granulocytes and T lymphocytes. The PNH clone size varied according to cell lineage, and clonal cells may obtain proliferative potential or gain a survival advantage compared with normal cells.
 XML Download
XML Download