

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hereditary gelsolin amyloidosis (HGA; also known as familial amyloidosis of the Finnish type, or FAF) is an autosomal dominant amyloidosis [1]. Clinical manifestations of HGA begin in the third or fourth decade of life with corneal lattice dystrophy, followed by cutis laxa and slowly progressive neuropathy [1234567]. Since the first report from Finland, additional cases of HGA have been identified in many ethnic groups [13456891011121314]. However, no Korean patients with HGA have been reported.
So far, four mutations in the gelsolin gene (GSN) have been identified as causes of HGA. The p.D214N mutation, the first discovered and the most common type, was identified in patients with typical presentations and complete penetrance [13456891013]. The second mutation discovered, p.D214Y, also leads to HGA that is clinically similar to that caused by p.D214N [56111214]. The p.D214N mutation was identified in Finnish, Japanese, American, Dutch, Portuguese, British, and Iranian families, and the p.D214Y mutation was found in Danish, Czech, and Brazilian families [13456891011121314]. It has been suggested that the p.D214Y mutation occurs irrespective of ethnicity, and that there are multiple founders [4713]. Recently, two additional mutations, p.G194R and p.N211K, were reported to be associated with renal amyloidosis in patients without corneal lattice dystrophy or neuropathies [121516]. We present the first report of a Korean family with HGA caused by p.D214Y mutation.
CASE REPORT
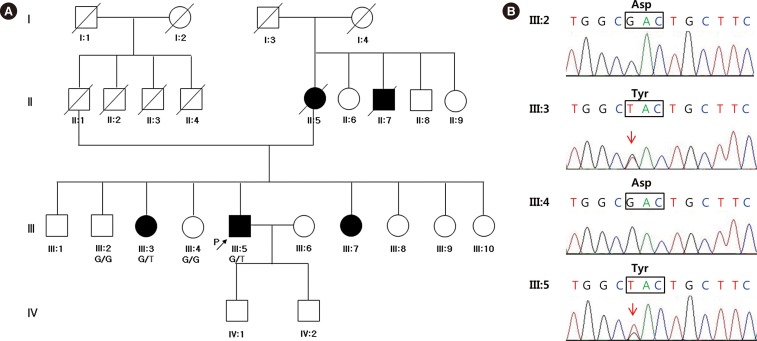
A 58-yr-old Korean male presented with involuntary facial twitching and weakness, loose skin, and a deep-furrowed tongue. The symptoms had been slowly progressing since he was 54 yr old. The patient's mother, maternal uncle, and two sisters suffered from the same symptoms (Fig. 1A). This Korean family has no non-Korean relatives. The proband complained of paresthesia of the right hand, intermittent cramps on the right calf, and decreased sweating on both hands. He had no ophthalmic complaints. Neurological examination revealed mild weakness in the orbicularis oculi and orbicularis oris muscles; however, no clear motor weakness or sensory changes were observed in the four extremities.
We performed electrophysiologic studies, including nerve conduction study (NCS), electromyography (EMG), and autonomic function tests (AFTs). In the facial NCS and EMG, reduced compound muscle action potential (CMAP) amplitudes and conduction velocity of the left facial nerve and reduced CMAP amplitude of the right facial nerve were identified. In addition, blink reflex tests showed borderline to mildly prolonged latencies of ipsilateral R1 and R2 and contralateral R2 waves with facial nerve stimulations on either side. Routine NCSs on the right extremities revealed reduced CMAP amplitude and conduction velocity and delayed terminal latency in the right peroneal nerve, and a mildly reduced distal sensory nerve action potential (SNAP) in the right median nerve. EMG of the right first dorsal interosseous muscle and tibialis anterior muscle revealed reduced recruitment of motor unit action potential. In AFTs, sympathetic skin responses to auditory, tactile, and electrical stimuli were missing in hands and feet, without evident abnormalities in heart rate variability, Valsalva ratio, Valsalva maneuver, or cardiovascular response to standing. Taken together, the electrophysiological studies demonstrated signs of bilateral facial nerve dysfunction, chronic denervation of the cervical and lumbar regions, and sudomotor dysfunction.
A trinucleotide repeat expansion analysis yielded no remarkable findings, excluding Kennedy disease. To identify the etiology of the undiagnosed neuropathy, we performed whole-exome sequencing (WES) following written informed consent.
Germline DNA was obtained from the four individuals (Fig. 1A). The exomes were targeted by using an SureSelect 50 Mb All Exon Kit (Agilent, Santa Clara, CA, USA) and sequenced on an HiSeq 2000 (Illumina, San Diego, CA, USA) (2×100 bp) according to the manufacturer's instructions. We generated an average of 78,148,365 reads resulting in an average of 6,989,878,321 high quality (≥MQ20) bases per sample. The average depth of the on-target regions was 90 fold. The reads were mapped to hg19 by using the Burrow-Wheeler Aligner (BWA 0.6.2.-r126, Wellcome Trust Sanger Institute, Cambridge, UK), and duplication removal and recalibrations were performed by using Picard Tools 1.64 (Wellcome Trust Sanger Institute) and GATK-Lite (Broad Institute, Inc, Cambridge, MA, USA), respectively. A total of 55,199 variants were called and functionally annotated by using ANNOVAR [1718]. To prioritize the variants, we applied the following criteria: allele frequency <0.01 in the 1000 Genome Project (http://browser.1000genomes.org/index.html) and the Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), and mutations predicted to have "deleterious" effects based on SIFT, Polyphen-2 LRT, Mutation-Taster, MutationAssessor, FATHMM, and GERP [18]. A total of 20 variants were present in the affected individuals but not in unaffected individuals. Among them, the p.D214Y mutation in GSN, a previously reported HGA mutation, was selected and validated by Sanger sequencing (Fig. 1B).
DISCUSSION
We present the first Korean family with HGA diagnosed by a family study and WES. Although the phenotypic manifestations of HGA were known to be typical, some variations have been reported [412151619]. A traditional, phenotype-first approach based on detailed clinical examination is inexpensive and readily available in the clinic. In clinically suspected cases, HGA can be diagnosed by verifying the presence of gelsolin amyloid in the endoneurium or skin. A phenotype-first approach, however, might allow HGA to go undetected in patients. Recently, WES has been successfully applied as a diagnostic tool in the identification of causative mutations in undiagnosed patients [20]. In spite of the diagnostic utility of WES, there are some drawbacks; for example, interpretation of the results can be challenging. To address this, a variant prioritization strategy based on a comprehensive annotation and segregation analysis can help to pinpoint causal variants. While a biopsy is invasive and may require a strong clinical indication, WES is less invasive and is a useful tool in a genotype-first approach.
After identification of the causative mutation, we were unable to confirm the phenotype, corneal lattice dystrophy, associated with this genotype, because the patient was lost to follow-up. However, other clinical manifestations and the results of the electrophysiologic studies were compatible with HGA. Despite the fact that no ophthalmic issues were presented and no eye exam was performed, WES successfully facilitated the clinical diagnosis of HGA. This is the first report of a Korean family with HGA, with cases genetically confirmed by WES.
 XML Download
XML Download