

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Germline mutations in the SET-binding protein 1 (SETBP1) gene (18q21.1) were first identified as causative genetic defects in children with Schinzel-Giedion syndrome (SGS), which is clinically characterized by severe mental retardation, distinctive facial features, and multiple congenital malformations [1]. In hematologic malignancies, recurrent somatic SETBP1 mutations have been recently reported in atypical chronic myeloid leukemia (aCML), unclassified myelodysplastic/myeloproliferative neoplasm (MDS/MPN), chronic myelomonocytic leukemia (CMML), and secondary AML (sAML) [2, 3]. SETBP1 encodes a protein that contains a homologous region to the SKI oncoprotein, a SET-binding region and three nuclear localization signals [4]. Piazza et al. [2] demonstrated that expression of a SETBP1 mutant showed a similar mechanism as SETBP1 overexpression. It has been suggested that overexpression of SETBP1 protects SET from protease cleavage [5]. Consequently, full length SET protein inhibits protein phosphatase 2 (PP2A) by forming a SETBP1-SET-PP2A protein complex, which leads to cell proliferation and expansion of leukemic cells [5].
In a recent report on SETBP1 mutations, the mutations in splicing factor 3B subunit 1 (SF3B1), U2 small nuclear RNA auxiliary factor 1 (U2AF1), and serine/arginine-rich splicing factor 2 (SRSF2), which are highly recurrent splicing pathway gene mutations in myeloid neoplasms, were investigated and a strong association between SETBP1 mutations and mutations in SRSF2 was demonstrated [6, 7].
Childhood AML is a rare and heterogeneous disorder, which is reported to occur in 7 cases per million children under 15 yr of age, and possesses nonrandom gene mutations contributing to the heterogeneity of disease [8]. Compared with adult AML, there are fewer studies on gene mutations in childhood AML. Here, we investigated the SETBP1 mutation and additional alterations of the splicing pathway genes SF3B1, U2AF1, and SRSF2 in childhood AML patients.
METHODS
1. Patients and samples
Fifty-three childhoods AML patients who were diagnosed and treated at Chonnam National University Hwasun Hospital from March 2004 to July 2013, were enrolled after obtaining Institutional Review Board approval and informed consent. Patient ages varied from 11 months to 17 yr, and the average was 10 yr. One patient was diagnosed with sAML and the other 52 were diagnosed with de novo AML. Clinical, cytogenetic, and molecular characteristics are shown in Table 1. Prognostic grouping of cytogenetically abnormal patients was based on conventional cytogenetic analysis and multiplex RT-PCR results [8].
Total DNA was extracted from bone marrow sample obtained at diagnosis by using a QIAamp DNA blood mini kit (QIAGEN, Hilden, Germany). Specimens were frozen in liquid nitrogen immediately after acquisition and were stored at -20℃ for further evaluation.
2. Direct sequencing of SETBP1, SF3B1, U2AF1, and SRSF2 genes
PCR and sequencing reactions were performed by targeting the hot spots of SETBP1 (exon 4, codon 778-979), SF3B1 (exons 14, 15, 16, and 18), U2AF1 (exons 2, 6, and 7), and SRSF2 (exon 1) by using primer pairs based on a modification of a published protocol [2, 9] (Table 2). Each amplified gene product was purified by using an AccuPrep PCR Purification Kit (Bioneer, Daejeon, Korea) and sequenced with a BigDye Terminator v3.1 Ready Reaction Kit (Applied Biosystems, Foster City, CA, USA). The gene sequences were compared by using the Blast2 program (http://www.ncbi.nlm.nih.gov/blast/bl2seq/bl2.htlm).
3. Cytogenetic and molecular analyses
Conventional cytogenetic analysis was performed on G-banded preparations from 48-hr bone marrow cell cultures. Balanced translocations were identified by multiplex reverse transcription (RT)-PCR using the HemaVision multiplex RT-PCR Screen Test kit (DNA Technology, Aarhus, Denmark) as previously described [10]. Mutation analysis of the fms-like tyrosine kinase 3-internal tandem duplication (FLT3-ITD) gene was performed by using PCR-restriction fragment length polymorphism (RFLP) and capillary electrophoresis for separation. The Nucleophosmin 1 (NPM1) gene mutation was evaluated by using Ipsogen NPM1-A and B/D Muta-Quant kit (QIAGEN) and the expression of Wilms tumor 1 (WT1) and brain and acute leukemia cytoplasmic (BAALC) genes was evaluated by using Ipsogen WT1 and BAALC Profile-Quant kit (QIAGEN) following the manufacturer's instructions.
RESULTS
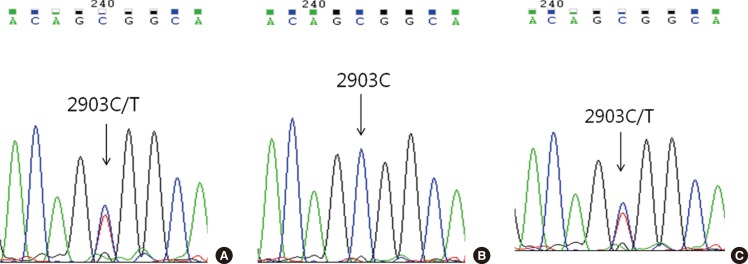
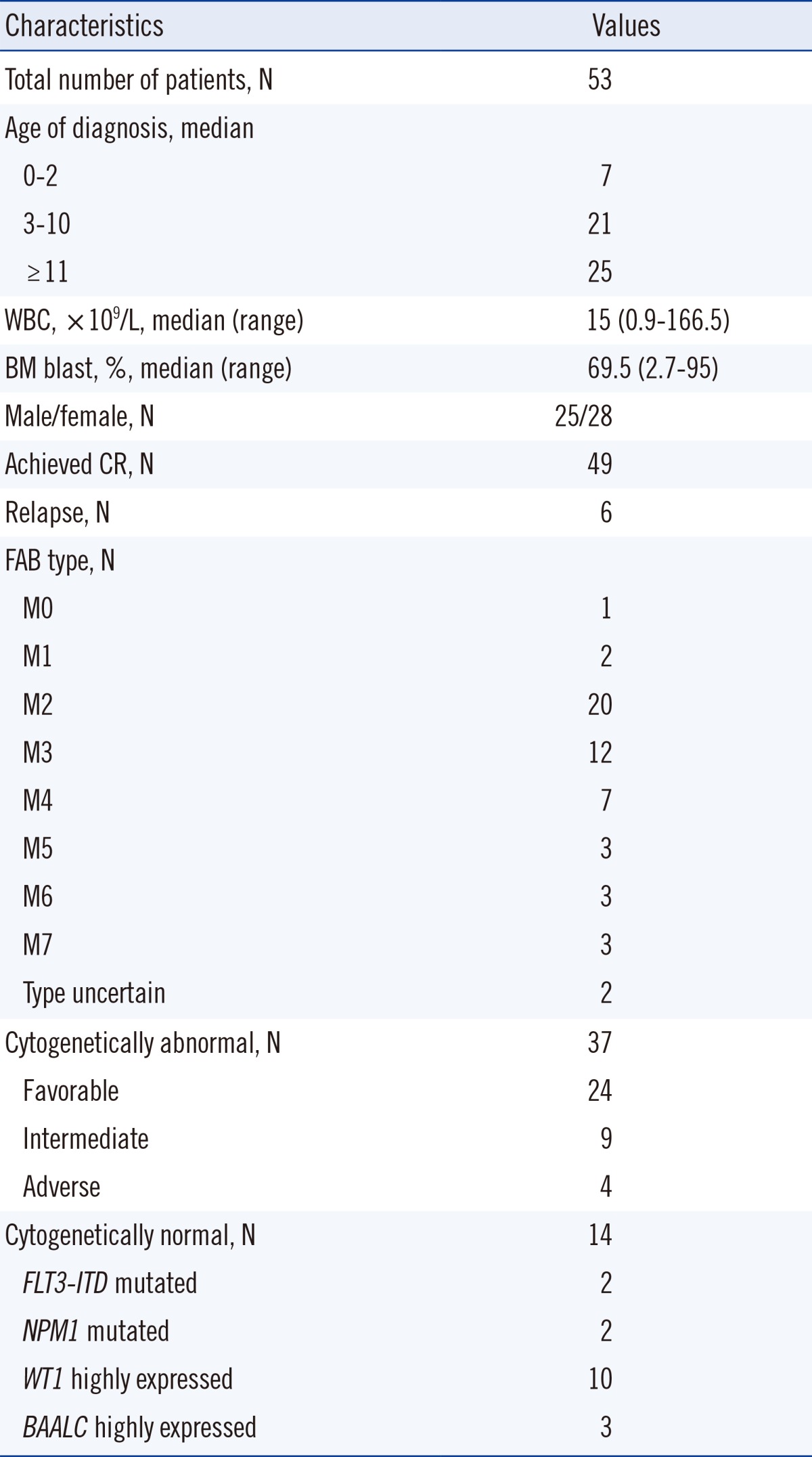
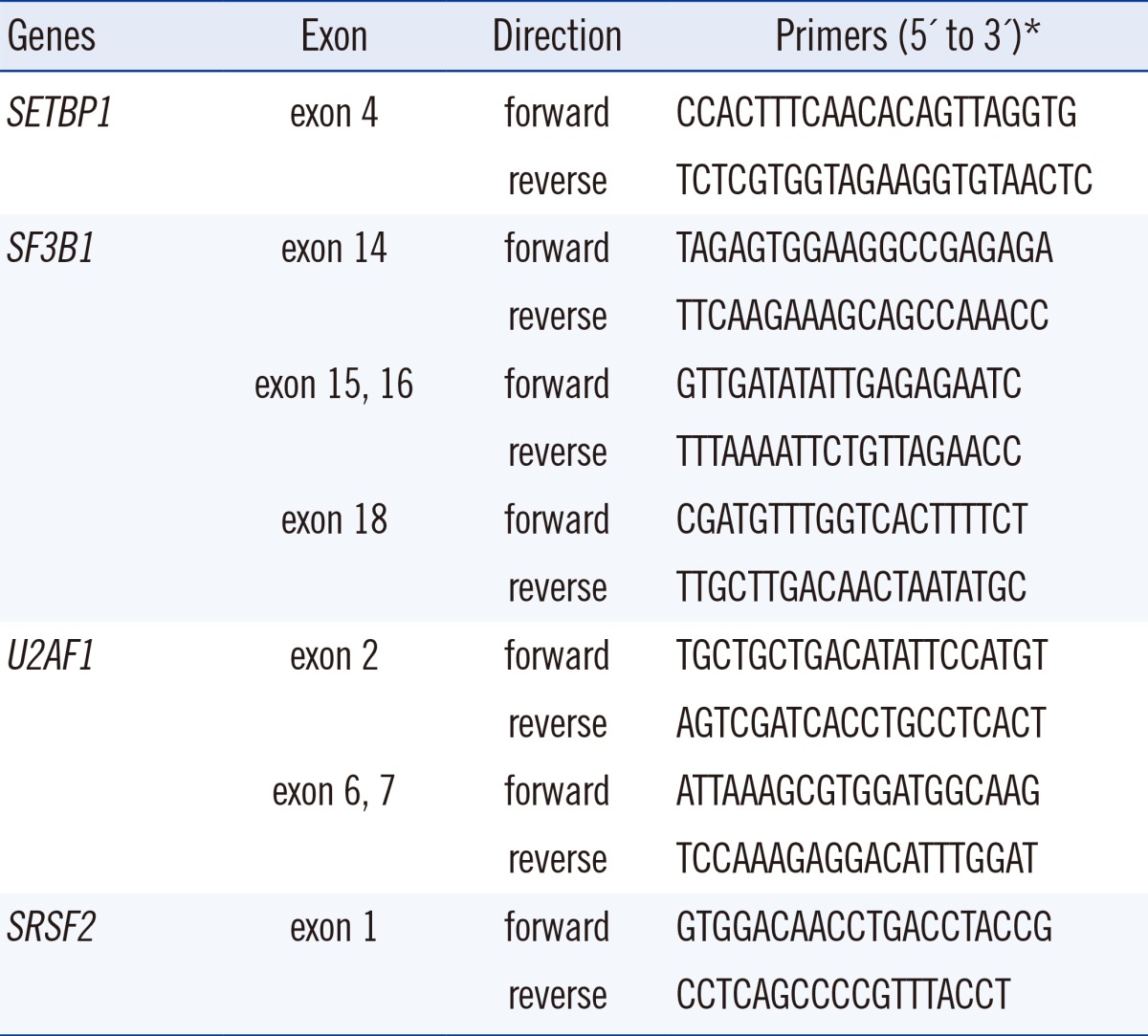
Childhood AML patients did not carry any of the recurrent SETBP1 mutations that had previously been identified in aCML or other related hematologic malignancies. However, one patient in the group of AML with RUNX1/RUNX1T1 rearrangement displayed c.2903C>T (synonymous) alteration in the mutational hotspot of SETBP1 (Fig. 1A). This patient's bone marrow samples were available for analysis from the initial diagnosis to the last follow-up, and we performed serial direct sequencing for the SETBP1 gene. On day 30 of the second induction chemotherapy, the SETBP1 gene alteration was undetectable coincident with the decrease of the RUNX1-RUNX1T1 transcript (Fig. 1B), but was present again during the consolidation therapy period (Fig. 1C).
None of the previously reported aberrations in the mutational hotspot of SF3B1, U2AF1, and SRSF2 were identified in any of the 53 pediatric AML patients. However, single nuclear polymorphisms (SNPs) were detected, with a particularly high frequency in SF3B1 and SRSF2. Forty-six (86.8%) patients showed SNP rs788018 in exon 18 of SF3B1 and all 53 (100%) patients showed SNP rs237057 in exon 1 of SRSF2. The genotype frequencies of SF3B1 SNP rs788018 were TT 13.2%, TC 39.6%, and CC 47.2%. The genotype frequencies of SRSF2 SNP rs237057 were CT 7.6% and TT 92.4%.
DISCUSSION
None of the 53 childhood AML patients showed a recurrent SETBP1 mutation found previously in MDS/MPN, MDS, sAML, or other related hematologic malignancies. Only one patient had a base-pair change in the hot spot of SETBP1 gene alteration. Since the number of alteration-positive cases was limited, it was not possible to compare the biologic or clinical differences between SETBP1-alteration positive and negative patients. However, the patient with a synonymous SETBP1 alteration showed the shortest survival time compared to the other patients with the same disease entity (median overall survival 4 months vs. 32 months, data not shown).
AML development is a multistep process and the leukemogenesis-altered proteins responsible for this clonal disease are presented as a "five-class model" [11]. This model comprises class I (signaling pathway components), class II (transcription factors), class III (epigenetic regulators), class IV (tumor suppressors), and class V (components of the spliceosome). Liang et al. [12] reported that the most frequent mutations in childhood AML were the class I mutations, and that epigenetic regulators (class III) mutations were much less frequent than they are in adult AML. In comparison of biologic properties and genetic abnormalities in childhood and adult AML, the frequency of de novo AML is higher (>95% vs. 83%) in childhood AML, but the frequency of sAML is lower (1% vs. 17%) in childhood AML [8]. In this study, similar results were observed. Among the 53 childhood AML patients, 52 patients (98%) had de novo AML, and we were unable to find any recurrent SETBP1 mutations. An analysis of 944 adult patients with AML and MDS did not detect any SETBP1 mutations in de novo AML patients and these results suggested that SETBP1 mutations play a role in the setting of MDS and sAML, but not in de novo AML [7].
A recent report by Shiba et al. [13] also demonstrated that the SETBP1 mutations are not recurrent in childhood AML. Considering the effect of a SETBP1 mutation in leukemogenesis, Damm et al. [3] compared the timing of SETBP1 mutations with other gene mutations during the evolution of CMML and sAML. Compared to spliceosomal gene mutations, SETBP1 mutations occurred after the SF3B1 and SRSF2 mutations. Therefore, the authors suggested that the SETBP1 mutations occur at later stages of disease evolution and might have more impact on the clinical course of the disease than on its initiation. Based on these reports, it is reasonable to propose that SETBP1 mutations have little impact on leukemogenesis in childhood AML.
The splicing pathway gene mutations, included in class V from the aforementioned five-class model, were identified as frequent somatic mutations of genes encoding multiple components of the RNA splicing machinery. Mutations in these genes were frequently detected in MDS and MDS-related disorders and were also identified in de novo AML. Yoshida et al. [6] found the SF3B1, U2AF1, and SRSF2 gene mutations in 4.6% of de novo adult AML, and Je et al. [14] in 5.6%. However, in this study, mutations were not found in SF3B1, U2AF1, and SRSF2 genes, indicating little relationship with childhood AML. Earlier studies on splicing pathway gene mutations in childhood leukemia reported similar results. Kar et al. [15] investigated SF3B1, U2AF1, and SRSF2 mutations in 49 patients with juvenile myelomonocytic leukemia, but none of the three splicing pathway genes was detected. Je et al. [14] showed that the SRSF2 gene was mutated in childhood acute lymphoblastic leukemia (1.5%), but not in childhood AML.
Synonymous SNPs are thought to have no effect on coding sequences, and therefore are not expected to change the function of the protein, in which they occur. However, studies on the impact on gene functions and the association with diseases are continuously reported [16, 17]. Recently, the prognostic significance of synonymous SNPs of WT1 (SNP rs16754) and IDH1 (SNP rs11554137) was evaluated in adult and childhood AML, and the results suggested a different outcome between SNP-positive and SNP-negative patients [18, 19, 20]. According to the SNP database [21], SNP rs788018 of SF3B1 appeared in 91.8% of the Japanese population and in 78.6% of the Han Chinese population. These results showed only a minor difference compared to the frequency (86.8%) of this study. SNP rs237057 of SRSF2 was found in almost 100% of the Japanese and Han Chinese populations, which was consistent with the present result. Reports have not yet been published regarding the clinical impact of a splicing pathway gene SNP.
In conclusion, although we found a synonymous gene alteration in one patient, childhood AML patients did not harbor recurrent SETBP1 mutations, and they did not harbor any mutations of SF3B1, U2AF1, and SRSF2 genes. Alterations of the SETBP1 gene or SFS3B1, U2AF1, and SRSF2 genes were not common genetic events in childhood AML, implying that these may not exert a driver effect in myeloid leukemogenesis during childhood.
 XML Download
XML Download