

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Suprasellar neoplasms include various types of tumors. The most common primary intracranial tumor involving the suprasellar mass is pituitary adenoma, which account for 10-15% of intracranial tumors [1]. However, many other types of tumors can manifest as a suprasellar mass, including not only primary intracranial tumors, but also metastatic brain tumors. Suprasellar tumors present with a variety of neurologic or endocrine dysfunctions depending on their site of origin and mass effect on adjacent structures [1].
With the exception of some cases such as germ cell tumors with elevated tumor markers, histopathologic exam of tumor tissue is required for a definite diagnosis. However, biopsy of the sellar area has substantial risks, so it is a challenge for physicians to decide whether to perform a biopsy of suprasellar mass.
Langerhans cell histiocytosis (LCH) is histiocytic disorder derived from myeloid progenitor cells that express CD34 surface antigen [2]. The clinical presentations of LCH vary depending upon the sites and extent of involvement. Common presenting symptoms include skin rash, dyspnea or tachypnea, polydipsia, and polyuria. LCH can involve nearly every organ, and commonly involved areas are bone, skin, and lymph nodes [3]. Because of its various manifestations, it is sometimes difficult to suspect LCH with only clinical findings.
Here we report a case of LCH, which manifested as a suprasellar mass with hypopituitarism and diabetes insipidus (DI), and was initially suspected as an intracranial germinoma. This case highlights the importance of histopathological diagnosis in patients with a suprasellar mass.
CASE REPORT
In June 2007, a 29-year-old woman presented with a 1-year history of polyuria and polydipsia. She also reported amenorrhea for 9 months. Her serum Na level was 139 mEq/L (normal range: 135-145 mEq/L), serum osmol was 302 mOsm/kg (normal range: 289-302 mOsm/kg), urine osmol was 67 mOsm/kg (normal range: 300-900 mOsm/kg), and the urine specific gravity was 1.005 (normal range: 1.005-1.030).
On hormonal evaluation, her prolactin level was elevated to 43.3 ng/mL (normal range: 2.8-29.2 ng/mL). Serum levels of follicle stimulating hormone (0.67 mIU/mL), luteinizing hormone (1.3 mIU/mL), and estradiol (54.0 pmol/L) were normal. Serum levels of free thyroxine was 3.6 pmol/L (normal range: 12-30 pmol/L) and thyroid stimulation hormone was 2.5 uIU/mL (normal range: 0.55-4.78 uIU/mL), suggestive of central hypothyroidism. She also showed adrenal insufficiency on the low dose ACTH stimulation test, with a peak cortisol level of 262 nmol/L (normal response: above 500 nmol/L). She was started on desmopressin, synthyroid, and hydrocortisone.
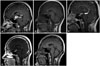
The brain magnetic resonance imaging (MRI) revealed a mass with a diameter of 2.3 cm on the suprasellar area, which showed strong enhancement. There was also a 1 cm-sized rimenhancing lesion in the pineal gland Fig. 1A. Her spine MRI revealed no abnormal leptomeningeal enhancement, and the cerebrospinal fluid (CSF) exam revealed no malignant cells. Her serum tumor markers were as follows: alpha-fetoprotein 1.4 µg/L (normal range: 0-20 µg/L), carcinoembryonic antigen 0.61 µg/L (normal range: 0-6 µg/L), beta-human chorionic gonadotropin 1.0 IU/L (normal range: 0-3 IU/L). CSF tumor markers were not examined.
A malignant intracranial tumor was suspected based on the above findings, and a biopsy of the brain lesion was recommended, but the patient refused. With a clinical impression of a germ cell tumor, three cycles of chemotherapy for advanced germ cell tumor (cisplatin+etoposide #3) was administered from June 11, 2007-August 25, 2007. In each cycle, cisplatin (20 mg/m2×5 days) and etoposide 300 mg/m2 (60 mg/m2×5 days) were given. After the chemotherapy, the tumor showed partial response Fig. 1B. Radiation therapy (whole ventricle 3,600 cGy/20 fractions, boost 900 cGy/5 fractions) was provided from October 11, 2007-November 4, 2007.
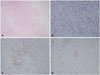
A follow up MRI on September 2008 showed increased mass size Fig. 1C. She was transferred to our hospital for further evaluation and management. Excisional biopsy was performed and the biopsy result showed LCH Fig. 2. Skeletal X-rays and bone scan revealed no bony lesions. No extracranial lesions were observed on chest CT and liver ultrasonography. Chemotherapy according to the LCH III protocol (for multifocal bone disease and special site involvement) was started from November 5, 2008. After the initial therapy (6 weeks with daily 40 mg/m2 oral prednisolone, and 6 mg/m2 i.v. vinblastine every 7 days), there was significant reduction in tumor size Fig. 1D. She received chemotherapy until July 15, 2010. She is now on regular follow up in our outpatient department. Due to panhypopituitarism and DI, she is now taking levothyroxine, hydrocortisone, and desmopressin, and we are considering starting sex hormone replacement therapy. She also has a visual field defect due to glaucoma and is taking glaucoma medication. Her visual acuity is only finger count possible for both eyes. She also complains of memory disturbance which inhibits normal social activities. The last brain MRI on June 2015 showed no new enhancing lesions or abnormal leptomeningeal enhancement, but atrophy of the pituitary gland was observed Fig. 1E.
DISCUSSION
LCH is diagnosed in all age groups but is most common in children from one to three years old. Its incidence is about three to five cases per million in children, and one to two cases per million in adults [4]. In adults, lung involvement is more common than in children, which is partly attributable to smoking [5]. On the contrary, lymphoreticular organ involvement occurs less frequently in adults than in children [4].
LCH involves the central nervous system (CNS) in about 6 percent of patients [3]. The most common manifestations of CNS involvement are diabetes insipidus and neurodegeneration. Diabetes insipidus presents with polydipsia and polyuria, and occurs secondary to infiltration of the posterior pituitary. Neurodegeneration is characterized by symptoms such as ataxia and cognitive dysfunction, and is caused by lesions of the cerebellum or the basal ganglia. Though rare, LCH can manifest as an intracranial mass lesion, usually in the hypothalamic pituitary area or the pineal gland [6].
In this case, the tumor was initially treated as a germinoma by clinical suspicion without a biopsy. Among pediatric CNS tumors, germ cell tumors accounts for 0.4-3.4 percent in western countries, and this incidence is higher and reported to be up to 11 percent of all pediatric brain tumors in Asian countries [7]. The most common lesion location is the pineal gland, and the second common is the suprasellar region. Surgery to obtain a tissue sample is highly recommended in patients with normal CSF and serum tumor markers to distinguish germinoma from other benign and malignant intracranial lesions, including LCH. Lesions other than germinomas have a higher chance of recurrence and need more aggressive therapy [8]. Stereotactically guided biopsy, with a 0 to 1.6% morbidity rate, provides a safe method of sampling tissues [9].
MRI is the study of choice for evaluating suprasellar tumors. In LCH, MRI reveals thickening of the pituitary stalk [10], and loss of the physiological hyperintense signal of the posterior pituitary on T1-weighted images in patients with DI. However, these findings can be observed in other CNS tumors such as germinomas [11]. Also, an enlarged pituitary gland is not pathognomic for germinoma, but is reported in about 8% of LCH patients [6]. Therefore, it is difficult to distinguish LCH from germinoma by means of MRI only.
Our case was initially suspected and treated as an intracranial germinoma. Neoadjuvant chemotherapy with cisplatin and etoposide was followed by radiation therapy. Previously, a phase II trial conducted by Buckner et al. [12] treated 17 CNS germ cell tumor patients aged 8 to 24 years with neoadjuvant chemotherapy (cisplatin and etoposide) followed by radiation therapy.
In this case, the LCH initially responded to chemotherapy for germ cell tumors (cisplatin+etoposide). Etoposide is known to be effective in the treatment of multisystem LCH [13], though this was not included in first line treatment of our case. Previously, a girl with a germinoma was reported who presented with DI and thickened pituitary stalk, who initially responded to LCH chemotherapy but showed rebound of tumor growth and was diagnosed as a germinoma after tumor resection [11]. Therefore, physicians should keep in mind that initial radiological response to inadequate therapy could be observed.
In conclusion, biopsy should be performed in patients with suprasellar tumors, even if there are clinical findings suggestive of a specific diagnosis. This case also highlights that we should suspect LCH in patients with DI, even in adults with no other symptoms of LCH.
 XML Download
XML Download