

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The first proton pump inhibitor (PPI), omeprazole, was launched in 1988. Because of its long duration of potent action and low side effect profile it quickly became the most frequently prescribed acid-suppressing therapy in the United Kingdom1 and throughout the world. In 2006 the first case of PPI-induced hypomagnesaemia and hypocalcaemia (PPIH)2 was reported, since when many case series have been published.3 In 2011 the United States Food and Drug Administration (FDA) issued a warning regarding the risk of hypomagnesaemia during long term PPI therapy. A review of the FDA adverse event reporting system indicated over 1,500 occurrences during the 5 years to 2009 and it is important to recognise that unlike many idiosyncratic drug reactions, PPIH is often delayed and may occur after several years on stable doses of therapy.4 The physiology of magnesium homeostasis is relatively poorly understood, with much of the focus has been on renal handling rather than intestinal absorption. This review considers the context and clinical factors associated with the PPIH.
PPIH PHENOTYPE
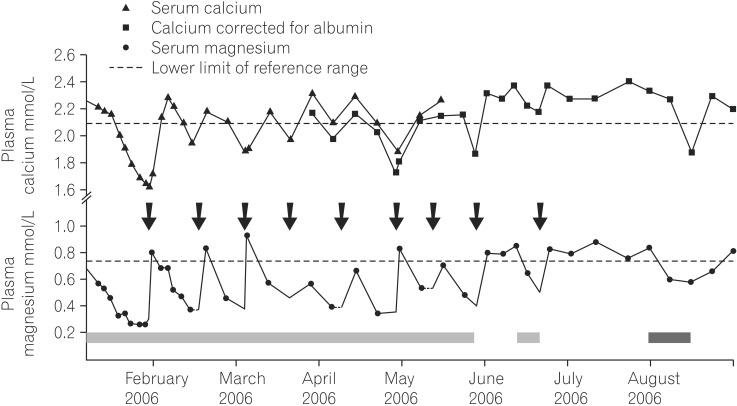
The first two cases of PPIH reported in 2006 described hypomagnesaemia and hypocalcaemia with low parathyroid hormone (PTH) levels.2 Since then over 65 cases have been published3 with varying symptoms and associated features. A systematic review of 36 cases5 reported that PPIH occurred after a median duration of 5.5 years therapy, with a range from 2 weeks to 15 years. The review also reported that PPIH resolves rapidly following discontinuation of PPI therapy and magnesium supplementation, within a mean 4.1 days. 95% of magnesium levels were in the reference range by 14 days. PPIH appears to recur rapidly in most patients within 4 days of re-challenge. These features have become the hallmark of the condition.
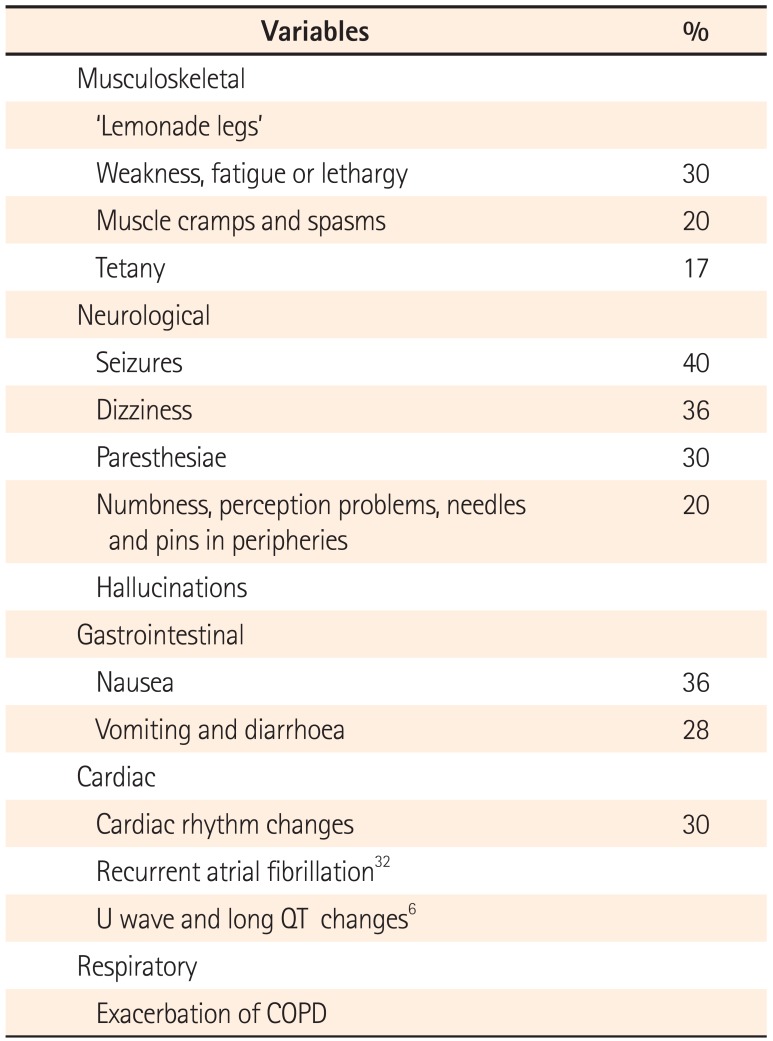
PPIH is associated with hypoparathyroidism, or inappropriately 'normal' PTH levels, related to low intracellular magnesium concentrations, which is involved in the PTH secretion cascade. Urinary magnesium excretion is appropriately low and has been reported to increase on cessation of PPI therapy.26 Fractional magnesium excretion and intravenous magnesium loading have been consistent with total body magnesium deficiency and appropriate renal handling.6 Coexistent hypocalcaemia and hypokalaemia7 are commonplace. Although Hess et al.5 reported moderate to severe hypocalcaemia in 70% of their cohort, they found no significant relationship to the magnesium nadir. Nonetheless, many of the presenting symptoms (Table 1) may be attributable to hypocalcaemia or combinations of electrolyte disturbances. Community studies8 suggest cases of mild hypomagnesaemia are under-recognised and it is likely so too are the severe effects, including fatal community cardiac arrhythmias.
Risk factors associated with PPIH in published series have included patents aged over 65 years,9 good adherence,10 concurrent diuretics and female gender.5 Not all factors have been replicated across cohorts, which is consistent with the variety of clinical presentations.11 Some debate has occurred over a wider association between PPI use and hypomagnesaemia. Cohorts in gastroenterology outpatients, coronary care units, dialysis units, medical inpatients and ambulatory community dwellers have attempted to address this issue. A systematic review of these series11 found significant heterogeneity between the studies, with no overall association between PPI use and hypomagnesaemia. However there was a weak association in hospitalized patients, or with a milder hypomagnesaemia definition of <1.8 mg/dL (0.74 mmol/L). Very high rates of hypomagnesaemia were seen in such series, ranging from 11-55% of PPI users and 4-53% of non-PPI users, raising questions about comparator groups.
Nevertheless, patients with additional susceptibility to hypomagnesaemia should be considered at higher risk of PPIH. Such patients include those with short bowel syndrome, or an inadequate magnesium or vitamin D intake due to malnutrition from any cause, alcoholism or anorexia, increased gastrointestinal (GI) magnesium loss from vomiting or diarrhoea, malabsorptive states including bypass surgery or enteroenteral fistulae and laxative abuse. Comorbitidies12 including diabetes mellitus, IBD, chronic pancreatitis, inherited renal pathology,9 renal tubular dysfunction, malignancy, hypertension and congestive cardiac failure13 are associated with hypomagnesaemia. The latter may be due to co-therapy with medications including loop or thiazide diuretics. Other drugs that may increase the susceptibility are epidermal growth factor (EGF) receptor modulators, platinum based chemotherapeutics,14 immunosuppressants including calcineurin and mammalian target of rapamycin (mTOR) inhibitors, aminoglycosides, amphotericin B and foscarnet,15 lithium, and digoxin,13 which have all been associated independently with hypomagnesaemia. Acute medical states such as the refeeding syndrome, after correction of diabetic ketoacidosis or metabolic acidosis, post-acute tubular necrosis polyuria and the 'hungry bone syndrome' are associated with acute drops in serum magnesium. Symptom onset can be insidious and a concomitant medical illness (such as sepsis or relapse of IBD) often appears to precipitate the presentation of symptomatic hypomagnesaemia. Indeed, so wide-ranging are both PPI prescription and these susceptibility factors that it is surprising that the condition is not more widely encountered or recognised.
It is notable that PPIH appears to be a class effect, with cases reported for all benzimidazole-substituted PPIs, but not H2 receptor antagonists (H2RAs). Different rates of events have been attributed to differing agent potency or market share,9 but the actual number of episodes is thought to far exceed reported cases.4 Consequently changing PPI within the class has not been effective treatment, although switching to an H2RA may alone be sufficient (Fig. 1). On the other hand, many series report that oral magnesium supplementation maintains plasma levels when continued use of a PPI has been necessary for the original indication.
MAGNESIUM HOMEOSTASIS
Magnesium is predominantly resident in the skeleton and muscle with 1% of total body magnesium found in the extracellular fluid. It is the second most common intracellular cation, found prominently in the nucleus, mitochondria and endoplasmic reticulum, mostly bound to ATP for which it is an obligate cofactor. Intracellular free magnesium therefore represents only 1-2% of total cellular magnesium.
The average daily intake of magnesium is 300 mg (12 mmol), mostly from green vegetables, nuts, cereals, milk and almonds which are a particularly rich source. Absorption of around half of this intake occurs throughout the GI tract; 22% in the jejunum, 56% in the ileum, and 11% in duodenum or colon.16 Digestive juices are rich in magnesium, excreting approximately 2 mmol/day, equivalent to one third that absorbed daily. Absorption can be increased in response to low oral intake17 or vitamin D and decreased during vitamin D deficiency, intestinal disease (villous atrophy, CD, or intestinal resection), or through saponification by free fatty acids in fat malabsorption states. Magnesium has a dense charge, so each atom tightly coordinates with 6-7 water molecules, thus having the highest hydration radius of all physiological cations.16 It must be stripped of water molecules by transporting proteins before it can be absorbed. The control mechanisms are not well understood, although there are both passive and active mechanisms for magnesium absorption.
Hypomagnesaemia is independently associated with a range of chronic conditions thought to be associated with inflammation and oxidative stress. Particularly in the elderly it is associated with hypertension, atherosclerosis, stroke and ischaemic heart disease, insulin resistance and type 2 diabetes mellitus, as well as markers of endothelial or platelet dysfunction.18 In the population-based Rotterdam study, mild hypomagnesaemia was common, occurring in 2% of the population and was associated with an increased all-cause mortality of 39% (hazard ratio, 1.06-1.81), after adjusting for standard cardiovascular risk factors.
The frequently delayed onset of PPIH suggests that compensatory mechanisms accommodate for magnesium malabsorption until a stage is reached at which decompensation occurs. Rapid recurrence on rechallenge with the causative agent would, in these circumstances, be predictable before any replenishment of total body magnesium stores. Compensatory mechanisms include the mobilization of magnesium and calcium from bone, which could conceivably account for the proposed association between long-term PPI therapy and osteopenia. Furthermore, vitamin D deficiency can reasonably be expected to exacerbate PPIH.
1. Intestinal Absorption
Magnesium is a divalent cation. Passive intestinal absorption of magnesium via paracellular channels appears to dominate at normal levels of dietary intake. Claudins are membrane proteins integral to tight junctions which occur in tissue-specific monomeric and heteromeric combinations. Claudin-2 and claudin-12 are divalent cation channels expressed in the intestines, while claudin-7 is a monovalent intestinal cation channel. PPI therapy has been demonstrated to decrease passive magnesium flux across Caco-2 monolayers (an intestinal epithelial cell line) and is associated with decreased claudin-7 and -12 expression.16 Further, omeprazole decreases the negative electric field strength within claudin-7 and -12 channels16 required to strip the bound hydration shell before it can pass through the claudin channel. Increased acid exposure to the apical membrane appears to rectify magnesium currents even in the presence of a PPI.16 The magnesium flux changes appear independent of the extracellular calcium sensing receptor within intestinal cell layers.16
Active intestinal absorption appears to occur via transcellular channels. Transient receptor potential cation channels of the Melastatin subfamily (TRPM) 6 and 7 have gained recent attention for their role in both renal and intestinal magnesium transport. These 'chanzymes' located in the apical membrane of epithelial cells, appear to have both channel and α-kinase enzyme functions. TRPM7 is ubiquitous and appears to activate both myosin IIA heavy chain and annexin 1, which suggests a role in cell growth, survival and cytoskeleton function.19 TRPM6 appears to modulate TRPM7 via cross-phosphorylation20 and co-expression of the TRPM6/7 heterodimer enhances trans-membrane currents. TRPM7 knock out mice suffer intracellular magnesium depletion and growth arrest, while in humans, inherited TRPM6 mutations are associated with both convulsions and a protein losing enteropathy 4-5 weeks after birth, the syndrome termed 'autosomal-recessive hypomagnesaemia with secondary hypocalcemia'.21 TRPM6 is sensitive to anti-endothelial growth factor (anti-EGF) agents, thiazides, insulin and oestrogens, but its function is strongly influenced by extracellular pH. mRNA expression of TRPM6 has been shown to increase with dietary magnesium deficiency and omeprazole (suggesting an adaptive response) throughout the colon, particularly in the caecum.3 This is particularly relevant to patents with an ileostomy, because any acquired deficiency in intestinal TRPM6 affects magnesium transport and is the likely explanation why hypomagnesaemia is common after ileocolic resection. An inability to upregulate TRPM6 in response to PPI therapy is then likely to account for magnesium malabsorption, which can be expected to have slowly cumulative effects when net absorption is insufficient to meet bodily needs, as demonstrated in US FDA modeling.22
2. Renal Handling
Renal Magnesium absorption follows similar principles to that seen in the intestine. The majority is reabsorbed within the thick ascending loop via passive flow through claudins, though in the kidney claudin-16 and -19 are significant.15 Active absorption occurs in the distal collecting tubule where TRPM6 was first identified and plays a key role within a negative electrical gradient. Channels maintaining this gradient, including the thiazide sensitive sodium/chloride cotransporter, affect magnesium absorption, as do alterations in TRPM6 promoters including EGF and 17β-estradiol, and thiazides or tacrolimus which decrease TRPM6 expression.23 Basolateral membrane transporters are not well understood but mutations in CNNM2 are associated with hypomagnesaemia, representing the first discovery in this area.24
POTENTIAL MECHANISMS OF PPI INTERACTION
Benzimidazole PPIs are lipophilic membrane-permeable weak bases which accumulate in the acidic canaliculi of parietal cells. The low pH activates intracellular metabolites from the pro-drug to an active form which becomes trapped intracellularly. Once protonated, it binds cysteine residues (in particular Cys831) in the gastric H+K+ATPase (gHK-α) before they become membrane bound. This accumulation in acid environments has been proposed as specific to parietal cells, but non-gastric HK-α enzymes are found in colon, skin, prostate and pancreas, thereby being further potential mechanisms of the effect.
PPI use is associated with increased expression of the non-gastric H+K+ATPase (cHK-α) in the distal colon and has been shown to decrease activity by 30%.25 TRPM6-mediated magnesium absorption is stimulated by extracellular protons, so PPI use may limit compensatory increases in colonic magnesium absorption.326
However esomeprazole use over one week has been shown to increase the amount of intestinal protons (H+ molecules) by 3.2 fold in the mid and distal small bowel.22 Furthermore, PPIs decrease pancreatic secretions by 85%,27 since both the gastric and non-gastric HK-α enzymes are found in pancreatic interstitial cells, as well as both the apical and basolateral membranes of pancreatic duct epithelium. The interaction between PPIs, pH and magnesium absorption is therefore in an equilibrium that can be disturbed by any variation from normal of the components.
Modeling of magnesium flux suggests that only a 1-5% decrease in daily magnesium absorption could cumulatively contribute to whole body depletion, consistent with the generally long exposure period before symptomatic PPIH.22 Increased or uncompensated GI losses have been proposed as a potential mechanism. Early radiolabeled magnesium challenge studies suggested intestinal secretion is a minimal component of magnesium homeostasis,28 but this has not formally been evaluated either in patients on PPIs, nor those with PPIH. Up-regulation of magnesium absorption when dietary intake is restricted, indicates a sensing mechanism, likely located in epithelial cells, that responds to environmental magnesium levels. There is evidence of both transcription and translational mechanisms29 raising the possibility of further unknown factors. Several relatively uncharacterized transporters are upregulated in the presence of restricted magnesium diets, including ancient conserved domain proteins (ACDP) associated with the rare urofacial syndrome,20 mammalian magnesium transporters (MMgT),30 the SLC41A1 transporter31 and the family of magnesium transporters known as NIPA (non-encoded in Prader-Willi syndrome) proteins.29 Their role and localization within the colon is unclear and are potentially unexplored targets for PPI interactions.
CONCLUSIONS
PPI-related hypomagnesaemia is a rare, but increasingly recognized clinical conundrum requiring acumen and a high index of suspicion to diagnose. One of our patients (Fig. 1) evocatively described his symptoms as 'lemonade legs'. The typical phenotype is an older patient on long term PPIs, often on concomitant therapy with diuretics and other comorbidity such as an ileocolic resection or ileostomy or an acute diarrhoeal illness. Presentations vary between severe and mild but identified cases to date are likely the 'tip of the iceberg' with mild cases and fatal community arrhythmias unrecognised. Hypomagnesaemia is associated with worse long term outcomes and a 40% increase in all cause mortality. The mechanism of PPIH has yet to be elucidated, but impaired intestinal absorption through PPI inhibition of paracellular claudin-mediated divalent cation channels or transcellular active transporter channels, appears pivotal. Whilst it may be overcome with magnesium supplementation it is a generic effect of PPIs and switching to other forms of acid suppression together with correction of vitamin D deficiency is appropriate.
 XML Download
XML Download