

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Barth syndrome [BTHS (MIM 302060)], first described as a mitochondrial disease affecting neutrophils as well as cardiac and skeletal muscles, is a rare X-linked recessive disorder characterized by cardiomyopathy (CMP), neutropenia, failure to thrive, skeletal myopathy and 3-methylglutaconic aciduria.1)2) BTHS is caused by loss-of-function mutations of the tafazzin (TAZ) gene located on chromosome Xq28. In the present report, we describe a Korean patient with BTHS, who had a novel de novo mutation in the TAZ gene. This is the first reported case of a BTHS patient with a de novo mutation in Korea.
CASE
A 13-month-old boy was referred to our hospital with intractable heart failure with dilated CMP, to evaluate the necessity of cardiac transplantation. He was born at full term with a birth weight of 3.0 kg. He had no siblings and had no family history of CMP or sudden death. At the age of 6 months, he had been admitted to another hospital because of lethargy, tachypnea with respiratory distress, and mild fever. Rhinovirus was isolated from a nasopharyngeal swab by real-time polymerase chain reaction. Chest radiography had shown marked cardiomegaly with pulmonary edema and echocardiogram had revealed a dilated left ventricle (LV) (LV end-diastolic diameter of 38.6 mm) with 30% ejection fraction (EF) and moderate mitral regurgitation (MR). Under the impression of dilated CMP due to acute myocarditis, the patient had been prescribed digoxin, furosemide, spironolactone and captopril and discharged from the hospital after stabilization of his hemodynamic status.
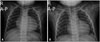
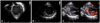
At presentation to our hospital at 13 months of age, the patient's body weight and height were 7.8 kg (< 3rd percentiles) and 72.5 cm (< 3rd percentiles), respectively. The characteristic facial features of BTHS were not observed. He could walk unaided and say "mama" and "papa". On physical examination, grade II/VI regurgitant systolic murmur was heard at the apex. Laboratory investigations revealed a white blood cell count of 2740/µL with an absolute neutrophil count of 438/µL. Serum concentrations of troponin I and lactate were 1.597 ng/mL (reference range: 0–0.3 ng/mL) and 3.8 mmol/L (reference range: 0.7–2.5 mmol/L), respectively. Serum electrolyte, sugar, liver function tests, creatine kinase (CK), and CK-MB levels were normal. The B-type natriuretic peptide level was 765 pg/mL. Urine organic acid analysis showed elevated levels of 3-methylglutaconic acid at 58.4 mmol/mol creatinine (reference range: 0–19 mmol/mol creatinine) and 3-methyglutaric acid at 7.5 mmol/mol creatinine (reference range: not detected). Chest X-ray revealed marked cardiomegaly with pulmonary edema (Fig. 1), and electrocardiogram showed normal sinus rhythm with left ventricular hypertrophy (Fig. 2). Echocardiography showed a dilated and hypertrophied spherical LV (LV end-diastolic diameter of 37.7 mm; z score: +4.6) with 23% EF, moderate MR, prominent endomyocardial trabeculations, LV non-compacted/compacted end-diastolic ratio of 2.3, color Doppler flow within the deep intertrabecular recesses and no prominent papillary muscle, confirming the diagnosis of dilated CMP with LV non-compaction (Fig. 3). At the age of 18 months, carvedilol was added, starting at 0.05 mg/kg/dose twice a day, and was gradually increased to 0.2 mg/kg/dose twice a day. No significant side effects of the medications were noted. After 1 year later from adding carvedilol, LV systolic function was remarkably improved (Fig. 3 and 4). Echocardiography showed a dilated LV with an EF of 53%, LV end-diastolic diameter of 35.6 mm (z score: +3.1), and mild MR (Fig. 4). During 2 years of follow-up, the patient had to be hospitalized twice for influenza infection and unspecified upper respiratory infection. Mild neutropenia persisted throughout the follow-up period (absolute neutrophil count of 150–650/µL). His characteristic clinical manifestations suggested BTHS. Gene sequence analysis revealed a hemizygous in-frame deletion of 9 amino acids within exon 10 of the TAZ gene, c.725_751del (p.Pro242_Glu250del). The mutation was not present in the Human TAZ Gene Mutation and Variation Database,3) and the variant was not detected in the patient's mother, indicating that it was a de novo mutation (Fig. 5). The patient was stable at his last outpatient visit at the age of 29 months. His body weight and height were 9.8 kg (< 3rd percentiles) and 81.5 cm (< 3rd percentiles), respectively. He could run well as well as ascend and descend stairs, but was still unable to speak beyond "mama" and "papa".
DISCUSSION
BTHS is a rare X-linked recessive disorder with clinical features of various types of CMP, neutropenia, skeletal myopathy, and 3-methylglutaconic aciduria. It has an estimated prevalence of 1 in 300000–400000 live births.1)3)4) The primary genetic defect in BTHS is mutations in the TAZ, which consists of 11 exons and is located at Xq28.5)6) This mutation lead to cardiolipin (CL) deficiency.2)7) The TAZ gene encodes TAZ with sequence homology to acyltransferases involved in phospholipid metabolism.8) TAZ is a mitochondrial protein located in the inner mitochondrial membrane and plays a crucial role in CL remodeling.2)8)9) CL is a predominant phospholipid of the mitochondrial inner membrane; it plays an important role in maintaining mitochondrial structure and is also involved in cellular apoptosis.7)10) Thus far, more than 200 different mutations in TAZ gene have been identified.2)3) Only 15% of boys carry de novo mutations that are not identified in their mother's somatic DNA.3) The correlations between the genotype and the phenotype have not yet been studied. There is no known racial or ethnic predilection for this mutation.6) However, the fact that the majority of BTHS patients have been reported from Europe, North America, and Australia suggests that the disease may be under-diagnosed.11)12) In Korea, one patient with BTHS was recently identified; he died after contrast dye injection for computed tomographic angiography.13) Our patient had a novel de novo mutation that was a hemizygous in-frame deletion in his TAZ gene, c.725_751del (p.Pro242_Glu250del), which was not inherited from his mother. To the best of our knowledge, this is the first reported case of a BTHS patient with a de novo mutation in Korea. The genetic analysis of this case was conducted by the help of the National Supporting Program for Genetic Diagnosis of Rare Diseases of the Korea Centers for Disease Control and Prevention. Infections due to neutropenia and cardiac complications are likely to be the major causes of morbidity and mortality in patients with BTHS.14) The survival rates of BTHS patients are gradually increasing mainly because of improved control of its complications such as heart failure and neutropenia.15) Early diagnosis of this condition is very important, since previous reports have shown that the mortality rate in patients who are identified prospectively and managed proactively decreases from 70% (retrospectively diagnosed patients) to just 10%.16) Despite this fact, a mean lag time between the first presentation and diagnosis of BTHS was 3.3 years.6) Our patient, too, was diagnosed after 12 months from the onset of symptom. He presented to us with acute circulatory collapse and respiratory viral infection. The patient was initially misdiagnosed with myocarditis/dilated CMP. The initial presentation of BTHS-associated CMP may mimic viral myocarditis/CMP or be precipitated by viral infection.6) His management was focused on controlling heart failure, and he showed a good response to medications including beta-blockers. Data from the literature suggest that heart failure can often be prevented by standard medical therapy in patients with BTHS, and the patients may remain stable for several years or may even improve over time like our patient, although most patients need to be maintained on standard cardiac medications for CMP.14)17) Our patient showed neutropenia at initial presentation, but this finding was mistakenly considered as the side effect of medications or bone marrow suppression due to frequent viral infections, because we thought that the initial diagnosis of acute myocarditis with proven etiology was definite. Therefore, BTHS should be included in the differential diagnosis of males presenting with CMP of apparent viral etiology, especially when neutropenia is present.6) Our patient's clinical course was unique in that he showed no gross motor delay over 2 years of age. He could walk unaided at the age of 12 months, unlike most patients with BTHS who achieve this milestone by the age of 2 years, and could run well at his last follow-up. Skeletal myopathy is widely recognized in BTHS, and most boys have at least mildly delayed gross motor milestones.6)14)
In conclusion, we identified a novel de novo mutation in the TAZ gene, c.725_751del (p.Pro242_Glu250del), which is the first genetically confirmed case of BTHS in a Korean male.



 XML Download
XML Download