

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Contrary to metastatic tumors of the omentum, primary tumors of the omentum are very rare and sporadically reported [1]. The pathologic spectrum of the omentum varies from lipoma, liposarcoma, leiomyoma, leiomyosarcoma, gastrointestinal stromal tumor, teratoma, fibrosarcoma, haemangiopericytoma, spindle cell sarcoma, desmoid tumor, fibroma, mesothelioma, etc. [2,3]. However, a primary malignant rhabdoid tumor in the omentum is very rare. Malignant rhabdoid tumors are highly aggressive tumors usually presenting in the central venous system, kidney and soft tissue in children. Herein, we present one case of primary malignant rhabdoid tumor of omentum in a 10-year-old girl for the first time.
CASE REPORT
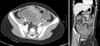
A 10-year-old girl visited the Emergency Department for abdominal pain. The pain had developed 7 days prior after getting hit with a ball. There were no associated symptoms such as nausea, vomiting, or diarrhea. On physical examination, the patient complained of tenderness in the lower abdomen. Laboratory findings showed that hemoglobin was 10.8 g/dL and C-reactive protein was 8.06 mg/dL. Other results were not remarkable. Ultrasonography showed a multiseptated large hematoma. A computed tomography scan was undertaken and showed a lobulating hemorrhagic tumor (9 cm × 6 cm × 6.5 cm) with hemoperitoneum in the lower abdomen and pelvis (Fig. 1). Initially, the mass was suspected as a hematoma resulting from bleeding of the lymphangioma or hemangioma in the mesentery. After 3 days of antibiotics treatment, her pain was relieved. For follow-up, we examined the mass by ultrasonography. It revealed a necrotic solid tumor enhancing with peripheral vessels and a small amount of hemoperitoneum. We assumed that the mass was a malignant tumor of the mesentery or retroperitoneum, and performed a laparoscopic exploration. There was a huge omental solid mass that was freely movable and free of adhesions to any other intra-abdominal organs. The mass was removed completely including remnant omentum via laparotomy. Several seeding nodules were found on the small bowel mesentery and Pouch of Douglas and were removed for pathologic confirmation.
Grossly resected multinodular solid masses showed irregular and infiltrative borders and the cut surface was tan gray and fleshy. There was multifocal hemorrhages and necrosis (Fig. 2). Microscopically nested round to polygonal tumor cells revealed infiltrative borders (Fig. 3A) and each tumor cell showed eccentric nuclei, prominent nucleoli, as well as a characteristic eosinophilic inclusion or globules in the abundant cytoplasm (Fig. 3B). These findings were consistent to that of "rhabdoid" cells. Immunohistochemically neoplastic cells expressed cytokeratin (Fig. 3C), vimentin (Fig. 3D), and epithelial membrane antigen. However, tumor cells showed negative reaction for desmin, smooth muscle actin and myosin. Importantly neoplastic cells revealed the absence of nuclear expression of INI 1 (Fig. 3E), which was recently discovered as a one of the most important histologic markers of malignant rhabdoid tumors [4].
Postoperatively, she recovered uneventfully and resumed her oral diet on the 3rd postoperative day. An fluorine-18 fluorodeoxyglucose positron emission tomography/computed tomography was undertaken and revealed no evidence of primary malignancy including kidney and central nervous system. At postoperative day 7, the patient began chemotherapy consisting of VDC/IE (VCR, Doxorubicin, Cyclophosphamide/Ifosfamide, Etoposide) regimen. Despite 4 cycles of chemotherapy of VDC/IE regimen, the mass in the Pouch of Douglas, where previously there had been documented seedings, was now apparent and showed peritoneal carcinomatosis through magnetic resonance imaging. She had been treated with intensive chemotherapy yet died after 9 months due to disease progression.
DISCUSSION
The omentum is a double layer of the peritoneum that encloses an organ and connects it to the abdominal wall. The greater omentum is a fat-laden fold of peritoneum that hangs down from the greater curvature of the stomach and connects the stomach with the diaphragm, spleen, and transverse colon. Because of its location and wideness, the greater omentum could be a common site for metastatic tumors from intra-abdominal organs. In contrast, primary tumors originating from the omentum are very rare. The omentum has abundant fat with connective tissues such as arteries, veins, and lymphatics. The omentum is lined by double-layered mesothelial cells with stroma containing fibroblast, pericytes, lipocytes, and lymphoreticular bodies [5]. It can lead to various primary tumors. Among them, common benign tumors known to develop are lipoma, leiomyomas, teratoma, gastrointestinal stromal tumors, and fibromas. The most common malignant lesions are leiomyosarcomas, hemangiopericytomas, and fibrosarcomas [5].
In general, the symptoms of omental tumors present as abdominal discomfort (45.5%), abdominal mass (34.9%), and abdominal distention (15.2%) [5]. Unfortunately, there are no specific findings differentiating the origin or nature of the mass in imaging studies due to the extent of the omentum and the adhered organs. A considerable finding is displacement of the stomach, the transverse colon and small bowel, by an extrinsic mass. As presented in our case, the origin of the mass could not be identified preoperatively, and it was confirmed by surgical exploration. The treatment of omental tumors are complete excision via omentectomy. We diagnosed the mass as a malignant rhabdoid tumor based on the typical cellular morphology and immunohistochemical stains.
Rhabdoid tumor of the kidney was identified as a variant neoplasm of Wilms' tumor in 1978 [6]. Malignant rhabdoid tumors mainly occur in the kidney, soft tissue and central nervous system, but tumors have been reported in tongue, nasopharynx, neck, mediastinum, thymus, heart, uterus, urinary bladder, vulva, skin, soft tissue, paravertebral region, liver, and gastrointestinal tract [6-8]. They occur either in infancy or early childhood and generally have a dismal prognosis compared to other pediatric cancers. The malignancy has a high tendency to metastasize early and outcome is poor despite surgery and chemotherapy. The published survival rates have ranged from 5 days to 5 months. The "rhabdoid" is thus named as it resembles a rhabdomyosarcoma microscopically although it does not show skeletal muscle markers [6-10]. The rhabdoid cells shows round to teardrop shape with vesicular nuclei and a single large nucleolus. There are ill-defined round to oval hyaline inclusions composed of intermediate filaments in cytoplasm [1]. Immunohistochemically, the rhabdoid cells express both cytokeratin and vimentin, but not myogenic differentiation nor INI1 protein [4,7,8]. The presence of a mutation of the hsNF5/INI1 gene located at chromosome 22q11 is helpful in establishing the diagnosis [6,9]. For treatment, an aggressive operation to achieve total resection is recommended, because the effectiveness of chemoradiotherapy has not been proven. The classic combination of ifosfamide, carboplatinum and etoposide is recommended, and multidrug protocols including topoisomerase I inhibitors are recommended [10].
Our patient showed a malignant tumor on the omentum with peritoneal seeding. The biology of rhabdoid tumors are, unfortunately, very aggressive. Rhabdoid masses in the omentum have the potential to spread diffusely and easily to the peritoneal cavity, which results in a poor prognosis, despite aggressive chemotherapy.
The diagnosis of tumors in the omentum is very difficult and requires confirmation by excision and histologic evaluation. A primary tumor originating in the ometum is rare but possible even as a malignant tumor. The prognosis depends mainly on tumor biology yet the malignancy of the omentum might cause severe outcomes because these masses can easily metastasize to mesentery and peritoneum tissues.


 XML Download
XML Download