

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cystinosis is a disease characterized by abnormal lysosomal cystine accumulation throughout the body, due to impaired lysosomal transport. It is a relatively rare condition, but may lead to fatal multiple organ failure caused by the systemic formation of cystine crystals. First reported by Abderhalden in 1903, cystinosis is an autosomal recessive disorder with an incidence of ~0.5 to 1.0 per 100,00012). Town et al.3) identified the causative gene, CTNS, encodes an integral membrane protein cystinosin, which has features similar to a lysosomal membrane protein, and CTNS mutations allow earlier confirmation of a cystinosis diagnosis.
In 2003, we reported a 12-year-old boy diagnosed with cystinosis the first case reported in Korea1). We began cysteamine (Cystagon, Orphan Europe SARL, Paris, France) treatment after confirming the diagnosis, and the patient was closely followed for additional complications. During the ensuing decade, we encountered various endocrine and nephrologic complications. After ensuring compliance with his prescribed therapy, the patient's serum leukocyte cystine levels were re-evaluated. Here, we present the therapeutic outcome of this cystinotic patient and describe his clinical course throughout adolescence and into early adulthood.
Case report
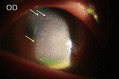
The patient was 12 years 8 months old when he first visited Yeouido St. Mary's Hospital, presenting with short stature, general weakness, and photophobia; his perinatal and family histories were unremarkable. At the time of diagnosis, the patient showed severe growth retardation with age-specific standardard deviation score (SDS) for height and weight of –6.3 and –3.2 and a prominent bone age delay (bone age, 2 years 6 months). Laboratory findings revealed chronic renal failure and primary hypothyroidism1) (Table 1). The patient had prepubertal testicles (2.5 mL) and an absence of pubic hair, but his neurocognitive function was not impaired. Slip-lamp examination confirmed the deposition of cystine crystals on his cornea and iris. A serum leukocyte cystine level assay (UCSD Cystine Determination Lab, San Diego, CA, USA) revealed 8.4 nmol half-cystine/mg protein (normal, <0.2 nmol half-cystine/mg protein)2) at the time of diagnosis. Cystinosis was made diagnosed based on the patient's ophthalmic manifestations and biochemical profile.
The patient started with 0.33 g/m2/day of oral cysteamine (Cystagon), which was gradually increased to 1.9 g/m2/day over the initial 4 weeks of treatment. Thyroid hormone replacement (2.0 µg/kg of levothyroxine) and routine ophthalmologic check-ups were also required as part of his therapy. However, the patient's overall therapy compliance was poor due to socioeconomic issues. At the age of 15 years, the patient was started on recombinant human growth hormone treatment (0.1 IU/kg/wk) and continuous ambulatory peritoneal dialysis. Growth hormone therapy continued until the age of 23 years when his bone age reached 16 years 5 months, and the patient showed good compliance. When the patient was 19-years-old, an ophthalmic cysteamine solution (Cystaran, Sigma-Tau Pharmaceuticals, Gaithersburg, MD, USA) became available, allowing combined topical (4.4 mg/mL of cysteamine, 1 drop/hr) and systemic therapy.
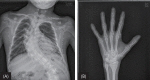
When the patient reached a chronological age of 26 years, his skeletal maturation was complete and he had achieved a final height and weight of 134 cm (SDS of –7.4) and 28 kg(SDS of –9.0) (Fig. 1). The patient's thyroid function had been normalized by levothyroxine administration. His testicular volume was 12 mL and he demonstrated abundant pubic hair; however, biochemical profiles revealed high levels of gonadotropins and sex hormone binding globulin (SHBG), with low androgen levels (Table 1). Peritoneal dialysis was unsuccessful at ameliorating the patient's deteriorating renal function, and he was scheduled for kidney transplantation. In addition, scoliosis developed and a hand X-ray revealed subperiosteal resorption indicative of renal osteodystrophy (Fig. 2). Further, continued visual deterioration resulted in keratoplasty when the patient was 22 years-old (Fig. 3). The patient had low self-esteem with demonstrated poor scholastic performance and social adaptability. The most recent serum leukocyte cystine measurement was 0.15 nmol half-cystein/mg protein (<0.1 nmol half-cystein/mg protein is optimal, but <0.2 nmol half-cystein/mg protein is beneficial); a cysteamine dosage of 1.5 g/m2/day helped reduce his serum cystine level without any side effects (Table 1).
Discussion
Although a favorable outcome is difficult to anticipate for cystinotic patients, cysteamine administration in conjunction with renal transplantation is the therapeutic maneuver most likely to increase the patient's quality and duration of life. Clinical presentation, biochemical profile, and ocular findings directed us to an early diagnosis of cystinosis, and led to the prompt intervention that was expected to minimize the irreversible sequelae. Nevertheless, in this patient, the clinical consequences during young adulthood were serious and disease progression continued. We speculate that the 2 major factors contributing to his poor long-term outcomes were older age at diagnosis and poor compliance during the initial treatment period.
Cysteamine bitartrate, currently the only available therapeutic drug, depletes intralysosomal cystine via a disulfide exchange reaction4). Even though the magnitude of the clinical manifestations may vary, major complications generally involve the kidneys, corneas, and some endocrine organs. The next most frequently affected sites are the bone marrow, spleen, and liver; neuromuscular abnormalities are common. The associated mortality and morbidity rates are generally high and are often manifest during adulthood. An evaluation of adult American cystinotic patients (median age at diagnosis was 24 months) showed that 7 out of 36 patients died between 18 and 34 years of age due to aspiration, pseudobulbar palsy, uremia, or unexplained sudden death4). Cystinosis has a direct and irreversible clinical course; therefore, its prognosis depends on the timing of cysteamine treatment2).
The most often reported endocrinopathy in adulthood is hypothyroidism, followed by primary hypogonadism5). Due to fibrosis and crystal accumulation in the testicular tissue6), biochemical profiles have demonstrated low serum testosterone and high SHBG (converted to 0.124 pg/mL of free testosterone7)) levels with elevation of serum luteinizing hormone and follicular stimulating hormone levels (Table 1). Although a typical cystinotic patient develops primary (hypergonadotropic) hypogonadism, the patient's sexual maturity progressed to midpuberty. To restore secondary sex characteristics and avoid infertility, routine gonadal function monitoring and cotreatment by urologists are initial steps required before considering testosterone replacement. By the end of the first decade of life, the vast majority of cystinotic patients develop fibrosis and atrophy of the thyroid gland due to cystine deposits in follicular cells, a condition that eventually leads to hypothyroidism8); our patient only required a small dose of levothyroxine to achieve a euthyroid state.
Severe growth retardation is a clinical hallmark of cystinosis, with only a few patients attaining a final height >150 cm2). Growth is interrupted not only by hypothyroidism, but also by proximal tubule dysfunction, causing substantial electrolyte loss and poor bone maturation aggravated by impaired calcitriol metabolism2). Such conditions may contribute to profound growth failure that cannot be restored using standard growth hormone treatment. Although the contribution of growth hormone therapy to the final height of cystinotic children is controversial, it is considered to be a treatment option without any guaranteed beneficial effect29). Growth hormone replacement, however, is not a fundamental treatment approach for cystinotic patients because of their compromised renal function. Pediatric endocrinologists emphasize that endocrinopathies or adult complications, such as hypogonadism and infertility, may adversely affect quality of life to the same extent as renal or ocular manifestations.
In our patient, the initial cysteamine dosage was gradually increased from 0.3 g/m2 to 1.5 g/m2, as recommended by international guidelines2). Our patient showed poor therapeutic compliance until he was 16-years-old (4 years after diagnosis) due to the high treatment cost of and an indifference towards the disease. Soon after compliance improved, the patient became punctual for all scheduled clinic visits. Compared with the maximum nontoxic dose of 1.95 g/m2910) his current cysteamine dose is reasonable. Health care providers should regularly check serum leukocyte cystine levels to monitor treatment response and the need for therapeutic dose adjustment. In our case, repeated testing, according to this recommendation, was not easy. Based on the most recent measurement of out patient's leukocyte cysteine levels, however, we can infer that the cysteamine level was maintained within a reliable range, following the patient's compliance improvement.
Future management will focus specifically on the potential hypogonadism, psychosocial support, and administration of an appropriate cysteamine dose following successful kidney transplantation. Therefore, the key approach for cystinotic patient is to establish an optimal treatment strategy through cooperative efforts among endocrinologists, nephrologists, and genetics specialists.
In conclusion, cystinosis is a rare genetic disorder that was once considered fatal, but is now treatable. Early detection and improved understanding of the disease should delay clinical progression towards irreversible complications. In the cystinotic patient, early administration of cysteamine followed by close monitoring of clinical and biochemical profiles and appropriate psychosocial support, are key to minimizing lifelong complications.

 XML Download
XML Download