

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
The 22q11.2 deletion syndrome, also known as DiGeorge syndrome, is a genetic disorder associated with a large number of clinical findings, including cardiac defects, abnormal facial morphology, thymic hypoplasia or aplasia, cleft palate, and hypocalcemia [1]. Approximately 75% to 80% of patients with a 22q11.2 deletion have congenital heart disease, especially conotruncal defects [2]. The most frequently reported are tetralogy of Fallot (TOF), followed by ventricular septal defect (VSD), interrupted aortic arch (IAA) type B, and persistent truncus arteriosus (PTA) [3].
Since fluorescent in situ hybridization (FISH) was first used to detect 22q11.2 deletion [4], many large studies have been conducted investigating the associated cardiovascular anomalies in pediatric or adult patients [5-9]. Because of the strong correlation of 22q11.2 deletion with cardiac defects, many obstetricians recommend routine genetic screening for 22q11.2 deletion by FISH when heart defects are diagnosed on prenatal ultrasound. However some cardiac diseases have rarely, or never, been reported in fetuses with 22q11.2 deletions [3].
As there have been few prenatal studies, we evaluated data collected over 10 years at a single institution to characterize the spectrum of prenatally diagnosed congenital heart disease in 22q11.2 deletion syndrome and to provide guidelines for prenatal screening.
Materials and methods
This retrospective study conducted at Asan Medical Center, Seoul, Korea, between September 2002, and December 2012. The institutional review board approved this study. During the study period, there were 1,137 fetuses that had prenatal screening for 22q11.2 deletions because of suspected congenital heart diseases. All fetuses received a detailed, high-resolution ultrasound examination including echocardiography. Examinations were performed using an Accuvix XG or XQ ultrasound scanner (Samsung Medison Co. Ltd., Seoul, Korea) with a 2 to 6 MHz transabdominal probe.
The maternal characteristics and presence of major cardiac anomalies, including TOF, VSD, IAA, PTA, double outlet right ventricle (DORV), coarctation of aorta (CoA), hypoplastic right heart, and associated vascular anomalies, such as right aortic arch (RAA), were recorded for each case with a confirmed 22q11.2 deletion. Parental screening for 22q11.2 deletion was also noted. All patients with a cardiac defect confirmed by prenatal ultrasonography were included in the analysis, even if lost to follow-up after diagnosis. The prenatal diagnoses were confirmed at autopsy or by postnatal echocardiography.
Conventional karyotyping and FISH analyses were performed using cells obtained from amniotic fluid or cord blood. Karyotypes were examined using G-banding. FISH analyses were performed using HIRA probes (Vysis Inc., Downers Grove, IL, USA), which map to 22q11.2 and an ARSA control probe, which maps to 22q13.
The prevalence of 22q11.2 deletion in the fetuses with cardiac defects and the frequency of each cardiovascular anomaly were demonstrated as percentages. The maternal age and gestational age at diagnosis were shown as a median and range.
Results
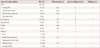
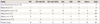
During the study period, a 22q11.2 deletion was confirmed in 53 of the 1,137 fetuses (4.7%) who were diagnosed with any congenital heart disease prenatally. The maternal characteristics of fetuses diagnosed with 22q11.2 deletion are demonstrated in Table 1 and the spectrum of cardiovascular diseases observed in the 53 fetuses is shown in Table 2.
1. Tetralogy of Fallot
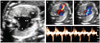
Almost half of the fetuses (24/53, 45%) showed TOF; simple TOF and TOF with major aorto-pulmonary collateral arteries (MAPCAs) being the most frequent. TOF was commonly associated with RAA (n=16, 67%). Three fetuses with absent pulmonary valve syndrome had severe dilatation of the main and branching pulmonary arteries that was associated with a RAA (Fig. 1). All of three fetuses were lost to follow-up after the diagnosis was made.
2. Interrupted aortic arch
Of the 10 fetuses (19%) with prenatally diagnosed IAA, eight were confirmed postnatally or postmortem, and all were type B. RAA was found in two fetuses. One was a full term male baby who underwent successful surgical repair of the aortic arch. The baby was doing well when seen at a 4-month follow-up visit.
3. Simple ventricular septal defect
Five cases of VSD occurred as an isolated intracardiac lesion; three of those were associated with RAA. Four fetuses presented with perimembranous VSD and one with subarterial VSD.
4. Double outlet right ventricle
Three cases of a Fallot-type DORV and one of a VSD type were detected. Two of the fetuses also had RAA. All parents of fetuses with DORV opted to terminate their pregnancies, and the prenatal diagnoses were confirmed at autopsy.
5. Coarctation of aorta
Coarctation of the aorta was reported in two twin pregnancies. After counseling about the prognosis, the parents chose selective feticide, and both of the healthy twins were delivered at near term. The autopsies were not allowed in terminated cases. The CoAs were not associated with RAA.
Discussion
Conotruncal cardiac anomalies are characteristic features of the 22q11.2 deletion syndrome. The introduction of fetal echocardiography made it possible to diagnose cardiac defects prenatally with high accuracy. Although many postnatal studies have described the incidence and pattern of cardiac anomalies in patients with 22q11.2 deletions, data from prenatal diagnosis in large series is less extensive (Table 3) [10-15]. Most prenatal series assessed the incidence of 22q11.2 deletion in fetuses with cardiac anomalies that were detected by ultrasound. A study published in 1997 identified three fetuses with 22q11.2 deletions among 26 pregnancies (11.5%) with cardiac diseases [10]. Manji et al. [11] reported a similar prevalence of 10.8%, i.e., five deletions among 46 fetuses with conotruncal cardiac anomalies. They recommended FISH analysis for fetuses with cardiac defects other than hypoplastic left heart and echogenic focus in the heart, which are not known to be associated with 22q11.2 deletion. Boudjemline et al. [12] and Volpe et al. [13] reported somewhat higher deletion rates (20.7% and 19.8%) than the previous series, while Moore et al. [14] and Bretelle et al. [15] reported lower rate (3.1% and 4.7%, respectively). Moore et al. [14] suggested that the wide variations in reported prevalence could be explained by different selection criteria for the types of cardiac defects. The studies that included all detected cardiac abnormalities reported lower prevalence of deletion, and those that included only conotrucal cardiac defects revealed higher prevalence. The 4.7% prevalence observed in this study, which included all anomalies that were detected in the series, is in line with the findings of those earlier studies.
To our knowledge, this study included the second largest number of confirmed 22q11.2 deletions. As in the other reports, the most prevalent disease was TOF, followed by IAA. However PTA was found in only one fetus (1.8%), which is a much lower prevalence than reported in other studies. A Korean multicenter study reported that only three of 190 patients (1.7%) with cardiac anomalies associated with 22q11.2 deletion had PTA [9], and a Japaneses study in 100 fetuses also showed a low, 2% prevalence of PTA [16]. A review of the cardiovascular anomalies associated with 22q11.2 deletion found that PTA was more prevalent in Western than in Asian counties [3]. We also recently reviewed the 12 fetuses with postnatally confirmed PTA, and among nine who underwent FISH analysis, none had a 22q11.2 deletion [17]. From these findings, we carefully suggest that ethnic differences could account for the reported differences in prevalence of cardiac defects.
Several cardiac defects occur rarely, or have not been reported, in patients with 22q11.2 deletion. Momma [3] found that tricuspid atresia, double-chambered right ventricle, left atrial isomerism, total anomalous pulmonary venous return, aortic valve stenosis, hypoplastic left heart, and double aortic arch were seldom associated with 22q11.2 deletion. They also noted some cardiac anomalies that have not been reported, including right atrial isomerism, single ventricle, corrected transposition of the great arteries, Ebstein's anomaly, and pulmonary atresia with an intact ventricular septum. None of the aforementioned anomalies were detected in our series, except for one fetus with aortic valve stenosis. In cases with rarely associated heart diseases, genetic screening for 22q11.2 deletion should be recommended only if other vascular anomalies or characteristic features associated with the deletion are detected.
During the study period, we found only two fetuses with 22q11.2 deletion had normal cardiac anatomy. The prevalence of congenital heart disease in fetuses with 22q11.2 deletion was 96% (53/55), which was much higher than the 75% to 80% reported in other series [18]. All study cases were referred to our tertiary center because of suspected cardiac defects, and therefore were not a random sample. It is also difficult to diagnose the deletion during fetal life in affected patients who are nearly asymptomatic. These factors contributed to the higher prevalence of deletion-associated cardiac disease in our series.
Many extracardiac abnormalities are associated with the 22q11.2 deletion syndrome, such as abnormal facial morphology, cleft palate, thymic hypoplasia, polyhydramnios and renal or skeletal anomalies [19]. Facial dysmorphia can be diagnosed by prenatal ultrasound. Typical facial features include hypertelorism, prominent nasal bridge, and micrognathia [19]. However, minor or subtle facial abnormalities are difficult to identify in the fetal period. Fetal thymus can also be reliably evaluated during echocardiography [20]. A hypoplastic or absent thymus should raise the suspicion of 22q11.2 deletion. Chaoui et al. [21] reported a high sensitivity (90%) for prediction of 22q11.2 deletion when thymic aplasia or hypoplasia was detected. Recently Li et al. [22] assessed the fetal thymic volume by 3D ultrasound, and concluded that volumetric assessment was a better indicator of normal fetal thymus development than 2D measurement, and may help in deciding when to test for 22q11.2 deletion.
Most 22q11.2 deletions occur de novo, with only 8% being inherited [23]. A previous Korean multicenter study noted an inheritance rate of 8.5% [9]. Although the majority of parents were lost to follow-up and only 11 were screened for 22q11.2 deletion, our series also found a prevalence of 9.1% (1/11). When one of the parents has a deletion on chromosome 22q11.2, the offspring have a 50% risk of inheriting deletion. Therefore prenatal genetic diagnosis should be performed for familial cases [19].
The primary limitation of our study was that a final diagnosis could not be confirmed in many cases because a large number of fetuses were lost to follow-up. However, among the confirmed cases, diagnostic accuracy was high (96%, 25/26). Only one fetus was prenatally diagnosed with PTA, but was confirmed to have TOF with MAPCA.
The variable type of congenital heart disease was demonstrated prenatally as previously reported. Conotruncal cardiac defects except transposition of great arteries showed a strong association with 22q11.2 deletion. When such anomalies are diagnosed by fetal echocardiography, genetic testing should be performed for detection of 22q11.2 deletion. In cases with rarely associated cardiac defects, other associated anomalies, such as thymic hypoplasia or aplasia, should be evaluated to rule out the 22q11.2 deletion. Once the deletion is detected, parents should be referred for genetic counseling to discuss the consequences for their baby. Parental deletion screening should also be recommended to identify inherited deletions.

 XML Download
XML Download