

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Microglial cells are the resident innate-immune cells in the brain, which play a physiological role of immune surveillance and host defense. However, it is generally accepted that excessive microglial activation and subsequent neuroinflammation lead to synaptic loss and dysfunction as well as neuronal cell death. These changes are involved in the pathogenesis and progression of several neurodegenerative diseases [1,2].
Microglia, in response to brain injury or immunological stimuli, produce excess nitric oxide (NO) and proinflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α), which are considered to contribute to neuronal cell death and neurodegenerative processes. Multiple signaling pathways are involved in the expression of these molecules. Protein kinases such as mitogen-activated protein kinases (MAPKs), protein kinase C (PKC) and phosphoinositide 3 kinase (PI3K)/Akt are involved, while nuclear factor κB (NF-κB) are recruited in microglial activation pathways [3,4].
In addition, the role of nicotinamide adenine dinuclelotide phosphate (NADPH) oxidase has been emphasized in the microglial activation process. The NADPH oxidase, as the major source of microglial reactive oxygen species (ROS), is an enzyme complex composed of various membrane and cytosol subunits depending on cell types. Nox1, Nox2 and Nox4 isotypes are expressed as the catalytic subunits of NADPH oxidase in microglia [5,6]. Nox2 (also known as gp91phox) is the most responsive in activated microglia [6]. Nox2-dependent NADPH oxidase consists of two membranebound subunits and four cytosolic subunits in microglia. Upon phosphorylation by specific kinases, the cytosolic subunits form a complex and translocate to the membrane to dock with the membrane subunits and activate NADPH oxidase complex. Then, the activated NADPH oxidase generates the superoxide anion (·O2-) by reducing O2, that can be converted to other ROS molecules [7,8]. A growing body of evidence has indicated that NADPH oxidase and NADPH oxidase-derived ROS are associated with MAPKs, Akt and NF-κB, and thereby play an important role in microglial activation and neuroinflammation [3,9,10,11].
Many compounds derived from plants have been widely investigated as candidates for medicinal application. It has been previously reported that dieckol (DEK) isolated from marine brown alga Ecklonia cava (EC) exhibits anti-inflammatory and antitumor activity as well as free radical scavenging activity [12,13]. In the central nervous system, DEK was shown to inhibit cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) in activated microglia following LPS stimulation [14]. However, the molecular mechanism underlying neuroprotective activity of DEK still remains to be elucidated.
Therefore, we further investigated here whether DEK inhibits microglial activation via ERK, Akt, and NADPH oxidase-mediated pathways in activated microglia. In addition, we found, using a neuron-microglia co-culture system and microglial conditioned media system, that DEK inhibits neuronal cell death following excess activation of adjacent microglial cells.
METHODS
Reagents
Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin and Alexa Fluor 488-conjugated goat anti-rabbit antibodies were obtained from Invitrogen (Carlsbad, CA, USA). LY294002, Wortmannin and diphenyleneiodium (DPI) were purchased from Calbiochem (La Jolla, CA, USA). Antibodies against extracellular signal-regulated kinase (ERK), phospho-ERK, Akt, phospho-Akt, nuclear factor κB p65 (NF-κB p65) and inhibitor of κBα (IκBα) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against p38, phospho-p38 and inducible nitric oxide synthase (iNOS) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody against TATA binding protein (TBP) was purchased from Abcam (Cambridge, UK). Antibody against gp91phox was purchased from BD biosciences (San Jose, CA, USA). Horseradish peroxide (HRP)-conjugated immunoglobulin G (IgG) antibody was purchased from Vector Laboratories (Burlingame, MA, USA). All the other reagents were purchased from Sigma (St. Louis, MO, USA), unless indicated.
Extraction and isolation of DEK
DEK was kindly supplied by BotaMedi, Inc. (Seoul, Republic of Korea). Briefly, the whole plant of marine brown alga Ecklonia cava was collected from the Jeju Island coast in the Republic of Korea. The dried Ecklonia cava powder was extracted three times with 70% aqueous ethanol (EtOH) and then filtered. The filtrate was evaporated at 50℃ to isolate the ethanol extract. After the EtOH extract had been suspended in distilled water, it was partitioned two times with n-butanol. The n-butanol fraction was evaporated in a vacuum, and was subjected to ODS column chromatography. The DEK compound was finally purified by LH-20 column chromatography and the purified DEK was then confirmed by comparing their mass spectrometry, 1H-nuclear magnetic resonance (NMR) and 13C-NMR data to those in the reported literature [15].
Cell culture
Primary microglial cells were prepared from cerebral cortices of 1-day-old Sprague-Dawley rats as previously described [16]. BV-2 microglia cells [17] were a generous gift from Dr. E. Joe (Ajou University, Republic of Korea). HT-22 neurons, an immortalized hippocampal neuronal cell line [18,19], were obtained from Dr. B.H. Lee (Gachon University of Medicine and Science, South Korea). Enhanced green fluorescent protein (EGFP)-transfected B35 rat neuroblasoma cells (ATCC, CRL-2754) [20] were used to establish a neuron and microglia co-culture system. BV-2 microglia, HT-22 neurons and B35 rat neuroblastoma cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin, and maintained at 37℃ in a humidified atmosphere of 5% CO2.
Neurotoxicity analysis of microglial conditioned media
Neurotoxicity analysis of microglial conditioned media has been well established in previous reports [21,22,23]. BV-2 microglial cells were pretreated with DEK for 6 h, then the culture supernatants were discarded to eliminate any direct effect of DEK on HT-22 neurons. The cells were washed and further incubated with lipopolysaccharide (LPS, 1 µg/ml) for 24 h in the absence of DEK. The used culture media were collected from the culture dishes and centrifuged to remove the detached cells. Then, the supernatants were used as microglial conditioned media. HT-22 neurons were treated with various groups of microglial conditioned media for 24 h and neuronal viabilities were assessed by MTT assay and phase contrast microscopy.
Neuron-microglia co-culture
EGFP-transfected B35 rat neuroblastoma cells (B35-EGFP) were added to BV-2 microglia-plated wells and co-cultured (2.5 : 1 ratio) at the density of 1.5×105 cells/well in 48-well plate, as previously described [3]. An aminoglycoside antibiotic G418 (1 mg/ml) was used to select EGFP-transfected cells containing a neomycin-resistant gene. BV-2 microglial cells were treated with DEK for 6 h and then, washed before adding B35-EGFP cells to eliminate direct effect of DEK on B35-EGFP neurons. In the absence of DEK, neuron-microglia co-culture system was stimulated by LPS for 24 h. The numbers of B35-EGFP neuronal cells were assessed by counting EGFP-positive cells under a fluorescent microscope (Olympus IX70, Tokyo, Japan). Images of five random fields per well were captured and counted.
MTT cell viability assay
MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] was used to examine the effect of DEK on cell viability, as previously described [2]. After treatment, 250 µl MTT (2 mg/ml) was added to each well and the mixture further incubated for 2 h. The liquid in each well was then aspirated and 500 µl dimethyl sulfoxide (DMSO) was added, mixed thoroughly on a shaker for 30 min. Absorbance was subsequently read at 540 nm using a microplate reader (Model 550, Bio-Rad, Hercules, CA, USA).
NO quantification
LPS-stimulated nitric oxide (NO) production was measured using the Griess reagent as previously reported [2]. Microglial cells were pretreated with DEK for 1 h and then stimulated with LPS (1 µg/ml) in the presence or absence of DEK for 24 h. The supernatants were mixed with equal amounts of Griess reagent. Samples were incubated at room temperature for 10 min and absorbance was subsequently read at 540 nm using a microplate reader (Model 550, Bio-Rad, Hercules, CA, USA).
Enzyme linked immunosorbent assay (ELISA)
The amount of pro-inflammatory cytokines released into the culture medium was measured using mouse IL-6 and TNF-α ELISA kits based on the quantitative sandwich enzyme immunosorbent technique. The assay was performed according to the manufacturer's protocol (Invitrogen, Carlsbad, CA, USA) and the absorbance was read at a wavelength of 450 nm using a microplate reader (Model 550, Bio-Rad, Hercules, CA, USA).
DPPH free radical scavenging activity assay
DPPH (1,1-diphenyl-2-picrylhydrazyl) is a stable free radical that has a deep violet color in solution, and it shows a strong absorption at 517 nm due to its odd electron. Upon reduction by an antioxidant, the absorption disappears and the resulting decolorization (yellow). Ten µl of DEK was added to 190 µl of DPPH (0.15 mM) and mixed vigorously. The mixture was incubated in the dark at room temperature for 2 h, and the absorbance was read at 517 nm using a microplate reader (Tecan, Sunrise, AT, USA).
Intracellular ROS measurement
DCF-DA (2',7'-dichlorofluo-rescein diacetate) is a non-fluorescent acetylated form which diffuses into cell. The esterases cleave the acetate groups on DCF-DA within the cells, and the resulting reduced probe DCFH is oxidized by ROS, yielding the fluorescent product polar 2',7'-dichlorofluorescein (DCF). BV-2 microglial cells were seeded in a 96-well tissue culture plate at a density of 2×104 cells/well containing 200 µl medium. After stabilization for 12 h, the cells were pretreated with DEK for 1 h and then stimulated with LPS (1 µg/ml) in the presence or absence of DEK for an additional 30 min at 37℃. Then, the cells were incubated in DCF-DA (50 µM, 30 min) and fluorometric analysis was performed with the excitation/emission wavelength (485 nm/535 nm) using a Perkin Elmer LS-5B spectrofluorometer (Becton Dickinson, Mountain View, CA, USA).
Cytoplasmic and nuclear protein preparations
Cells were harvested from the culture dishes with trypsin and centrifuged at 12,000 ×g for 5 min at 4℃. Cytoplasmic and nuclear protein preparations were performed as previously described [2]. Briefly, cell pellets were resuspended in ice-cold hypotonic buffer [10 mM Hepes (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT), 10 µg/ml aprotinin, 0.2 mM phenylmethylsulfonyl fluoride (PMSF)]. After 10 min, the cytoplasmic extract was obtained as supernatant by centrifuging for 5 min. Nuclear pellets were obtained using ice-cold high-salt buffer [20 mM Hepes (pH 7.9), 20% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, and 0.2 mM PMSF].
Western blot analysis
Western blot analysis was performed as described [2]. The membranes were probed with the following primary antibodies: ERK and phospho-ERK (1 : 1000), p38 and phospho-p38 (1 : 1000), Akt and phospho-Akt (1 : 1000), gp91phox (1 : 1000), iNOS (1 : 1000), NF-κB p65 (1 : 1000), IκBα (1 : 1000), TBP (1 : 1000) and β-actin (1 : 5000).
RESULTS
Dose determination for DEK treatment
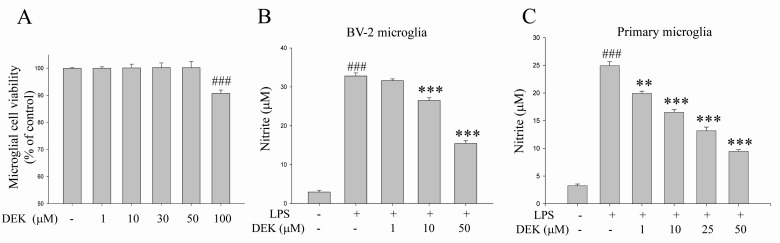
Dose of DEK treatment was determined based on cytotoxicity data and dose-dependency data (Fig. 1). BV-2 microglial cells were treated for 25 h with different concentrations of DEK. The cytotoxic dose of DEK was examined in BV-2 microglial cells using MTT assay (Fig. 1A). DEK did not show any toxicity at concentrations less than 100 µM. Therefore, experiments were accomplished at a concentration of 50 µM of DEK on the basis of cytotoxicty data (Fig. 1A) and dose-dependency data in BV-2 microglial cell line and primary microglia, respectively shown in Fig. 1B and 1C.
To assess the effect of DEK to inhibit NO release in LPS-stimulated BV-2 cell line and primary microglia, we measured the amount of NO in the culture medium using the Griess reagent. DEK pretreatment was carried out for 1 h prior to LPS stimulation. Then, microglial cells were treated with LPS (1 µg/ml) in the presence of DEK for 24 h. LPS treatment evoked robust NO release both in primary microglia and BV-2 cell line, and it was effectively suppressed by DEK (10~50 µM) treatment in a dose-dependent manner by 21.1±0.7% (10 µM) and 58.1±0.7% (50 µM) in BV-2 microglial cell line (Fig. 1B) and by 38.8±0.5% (10 µM), 54.2±0.7% (25 µM) and 71.3±0.3% (50 µM) in primary microglia (Fig. 1C), respectively.
Neuroprotective effects of DEK against microglial conditioned media-mediated neuronal cell death
It has been previously reported that the release of NO, IL-1β, and TNF-α is significantly reduced by DEK treatment in LPS-stimulated microglial cells [14]. We also observed these inhibitory effects of DEK in primary microglia and BV-2 cell line (Fig. 1B and C for NO, data not shown for IL-6 and TNF-α). Therefore, we investigated whether DEK protect neurons against microglia-mediated neuronal cell death both in microglial conditioned media system (Fig. 2A) and in neuron-microglia co-culture system (Fig. 2B). These two experimental models have been well established to determine microglia-mediated neurotoxicity in current days [3,21,22,23].
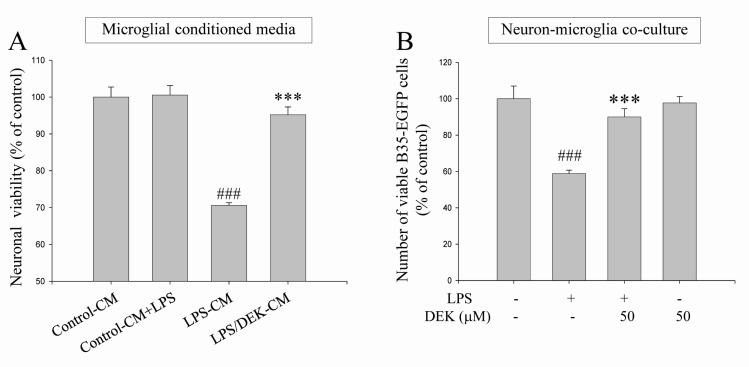
BV-2 microglial cells were pretreated with DEK (50 µM) for 6 h. Then, the culture supernatants were discarded to eliminate direct effect of DEK on HT-22 neurons. The microglial cells were further incubated with LPS (1 µg/ml) for 24 h in the absence of DEK. The conditioned culture media from BV-2 microglia were collected. Then, HT-22 neurons were treated with various groups of microglial conditioned media for 24 h and neuronal viabilities were assessed by MTT assay and phase contrast microscopy. Briefly, four microglial conditioned media groups were set up: (1) the conditioned media from control BV-2 cells (Control-CM); (2) the conditioned media from LPS-treated BV-2 cells (LPS-CM); (3) the conditioned media from BV-2 cells stimulated with LPS after DEK pretreatment (LPS/DEK-CM); and (4) LPS was added to the conditioned media from control BV-2 cells (Control-CM+LPS).
We found that treatment of LPS-CM markedly decreased cell viability of HT-22 neurons to 71.0±0.3% (Fig. 2A), compared with control conditioned media. Neuronal processes of most HT-22 cells treated with LPS-CM appeared to be shrunken, compared with control CM (data not shown). Cell viabilities of HT-22 neurons treated with LPS/DEK-CM were markedly increased, compared with that of LPS-CM-stimulated HT-22 neurons. These results suggest that the neurotoxic effects of LPS-CM were almost blocked by DEK pretreatment. On the other hands, addition of LPS into control CM (Control-CM+LPS) did not affect cell viabilities of resting neurons. Thus, it is indicated that HT-22 neurons are not be directly damaged by LPS.
Neuroprotective effects of DEK against microglia-mediated neuronal cell death in a neuron-microglia co-culture system
BV-2 microglial cells were pretreated with DEK (50 µM) for 6 h and then washed before adding B35-EGFP cells to eliminate direct effect of DEK on B35-EGFP neurons. Thereafter, B35-EGFP neurons were added to BV-2 microglial cells-plated wells. In the absence of DEK, neuron-microglia co-culture system was stimulated by LPS for 24 h. The numbers of B35-EGFP neuronal cells were assessed by counting EGFP-positive cells under a fluorescent microscope. These EGFP-transfected B35-EGFP neuroblastoma cells are useful to distinguish neurons from microglia in neuron-microglia co-culture due to their fluorescence.
The cell numbers of fluorescent B35-EGFP neurons were significantly decreased to 58.83±1.8% in this neuron-microglia co-culture system after LPS stimulation, compared to unstimulated control co-cultures. These results suggest that B35-EGFP neurons might be damaged by secretory molecules from adjacent BV-2 microglia while BV-2 cells in neuron-microglia co-culture system were stimulated by LPS. DEK-pretreated microglia did not affect neuronal cell viability without LPS stimulation. The cell viabilities were not affected by LPS alone in B35-EGFP neurons (data not shown). DEK pretreatment of microglia markedly blocked the cell death of B35-EGFP neuroblastoma cells to 90.0±4.6% after LPS stimulation in the neuron-microglia co-culture system (Fig. 2B), suggesting that DEK pretreatment has a beneficial effect for the cell viability in B35-EGFP neurons against LPS stimulation in the co-culture system. DEK pretreatment of microglia itself does not affect the cell viability in B35-EGFP neurons without LPS stimulation. These results, based on both microglial conditioned media system and the neuron-microglia co-culture system, indicate that DEK might have a therapeutic potential via attenuating microglia-mediated neurotoxicity implicated in some of neurodegenerative and neuroinflammatory diseases.
Inhibitory effects of DEK on phosphorylation of ERK in LPS-stimulated microglia
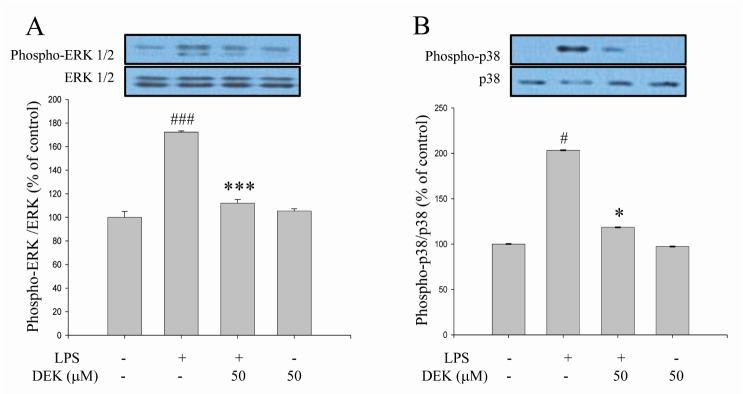
The subsequent experiments were performed to investigate the modulatory effects of DEK on the signaling pathways of microglia activation. We examined the effect of DEK on the LPS-induced phosphorylation of MAPKs such as ERK and p38 kinases in BV-2 cells using Western blot analysis. Following DEK pretreatment for 1 h, BV-2 microglial cells were stimulated with LPS (1 µg/ml) in the presence of DEK (50 µM) for 30 min. DEK significantly inhibited the phosphorylation of ERK-1/2 (Fig. 3A) and p38 (Fig. 3B) by 83.4±3.1% and 82.1±0.1% respectively. DEK did not affect LPS-induced phosphorylation of c-Jun N-terminal kinase (JNK) (data not shown). Our data are different from previous report that ERK phosphorylation was not affected by DEK treatment in LPS-stimulated microglia [14]. These discrepancies might be attributed to differences of experimental methods including dose and duration of DEK treatment.
Inhibitory effects of DEK on phosphorylation of Akt in LPS-stimulated microglia
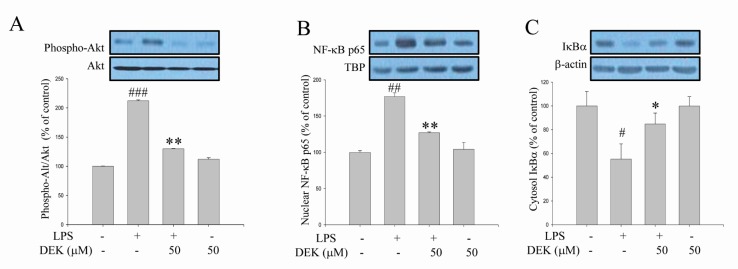
Akt phosphorylation is known to be involved in the degradation of IκB through IκB kinase (IKK) phosphorylation, and Akt/IKK/NF-κB signaling pathways are critical for LPS-stimulated microglial activation [9]. Although DEK was shown to inhibit NF-κB activation in LPS-stimulated microglia [14], Akt involvement has not been investigated yet. Thus, we investigated the effect of DEK on LPS-induced Akt-mediated pathways using Western blot analysis. After DEK pretreatment for 1 h, BV-2 microglial cells were stimulated with LPS (1 µg/ml) in the presence of DEK (50 µM) for 30 min. As shown in Fig. 4A, phosphorylation of Akt was markedly increased 30 min after LPS stimulation in BV-2 microglia, and DEK treatment significantly inhibited Akt phosphorylation by 73.31±0.5% (Fig. 4A). In addition, DEK treatment markedly attenuated nuclear translocation of NF-κB p65 by 64.7±1.1% (Fig. 4B) and degradation of IκB by 66.0±9.3% (Fig. 4C), similar to the previous report [14]. Taken together, we have shown that DEK effectively inhibits LPS-induced Akt phosphorylation and NF-κ B activation, suggesting that DEK may suppress microglial activation via Akt/IKK/NF-κB signal cascades.
Inhibitory effects of DEK on gp91phox expression and NADPH oxidase/ROS pathway in LPS-stimulated microglia
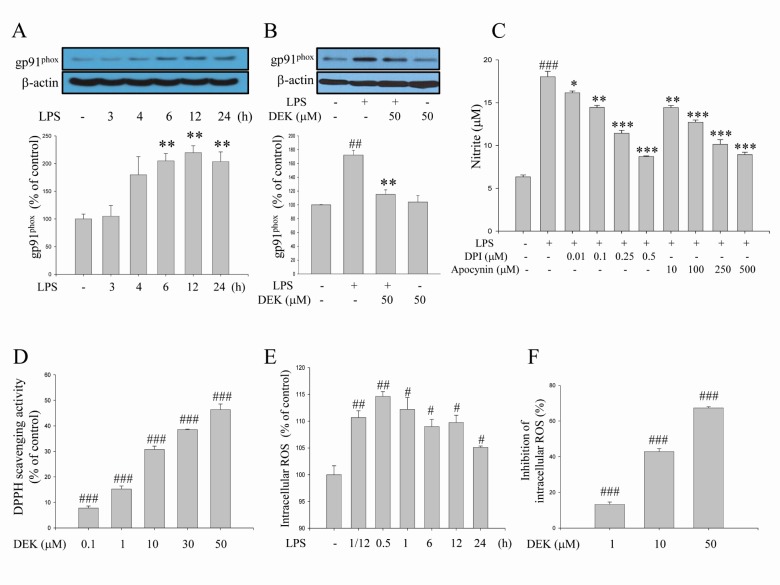
NADPH oxidase is the major source of ROS production in activated microglia. NADPH oxidase-derived ROS has been evaluated as a crucial component in the microglial activation processes. Nox1, Nox2 and Nox4 isotypes are expressed as the catalytic subunits of NADPH oxidase in microglia [5,6]. Nox2 (also known as gp91phox) is the most responsive in activated microglia [6]. It has been well established that expression of the gp91phox subunit was up-regulated in activated microglia [11,24,25].
We examined here the time course of LPS (1 µg/ml)-induced expression of the gp91phox component (Fig. 5A) using Western blot analysis. The increases in gp91phox expression were significantly shown 6 h after LPS stimulation. The increased expression was maintained through 24 h. The inhibitory effect of DEK on gp91phox expression was investigated after 6 h, based on the time course shown in Fig. 5A. The results indicated that DEK (50 µM) significantly blocked the LPS-induced increase of gp91phox expression in activated microglia (Fig. 5B). In addition, we applied apocynin and diphenylene iodonium (DPI), well-known NADPH oxidase inhibitors, to investigate whether NADPH oxidase regulates the secretion of pro-inflammatory mediator NO, which is a crucial event of microglial activation, in LPS (1 µg/ml, 24 h)-stimulated BV-2 microglia. As shown in Fig. 5C, apocynin and DPI dose-dependently inhibited NO production, indicating that NADPH oxidase is crucial in microglial activation pathway.
In addition, we observed that DEK exerts ROS scavenging activity (Fig. 5D), which is consistent with the previous report [12,14]. The time course of LPS-induced intracellular ROS levels indicates that the level of the intracellular ROS increased within 5 min, and maximized at 30 min and sustained until 24 h in LPS (1 µg/ml)-stimulated BV-2 microglia (Fig. 5E). Based on these time course data, the effects of DEK on LPS-induced ROS levels were elucidated 30 min after LPS treatment. After DEK pretreatment for 1 h, BV-2 microglial cells were stimulated with LPS (1 µg/ml) in the presence of DEK (1, 10, and 50 µM) for 30 min. We found that DEK significantly reduced intracellular ROS levels in dose-dependent manner in LPS-stimulated microglia (Fig. 5F).
Taken together, Fig. 5 indicated that DEK prevented LPS-induced increase of both intracellular ROS levels and NADPH oxidase component gp91phox expression levels. The results suggest that DEK suppresses NADPH oxidase-ROS pathway through both scavenging intracellular ROS and suppressing gp91phox expression.
Neuroprotective effects of the blockade of PI3K/Akt and NADPH oxidase against microglia-mediated neuronal cell death
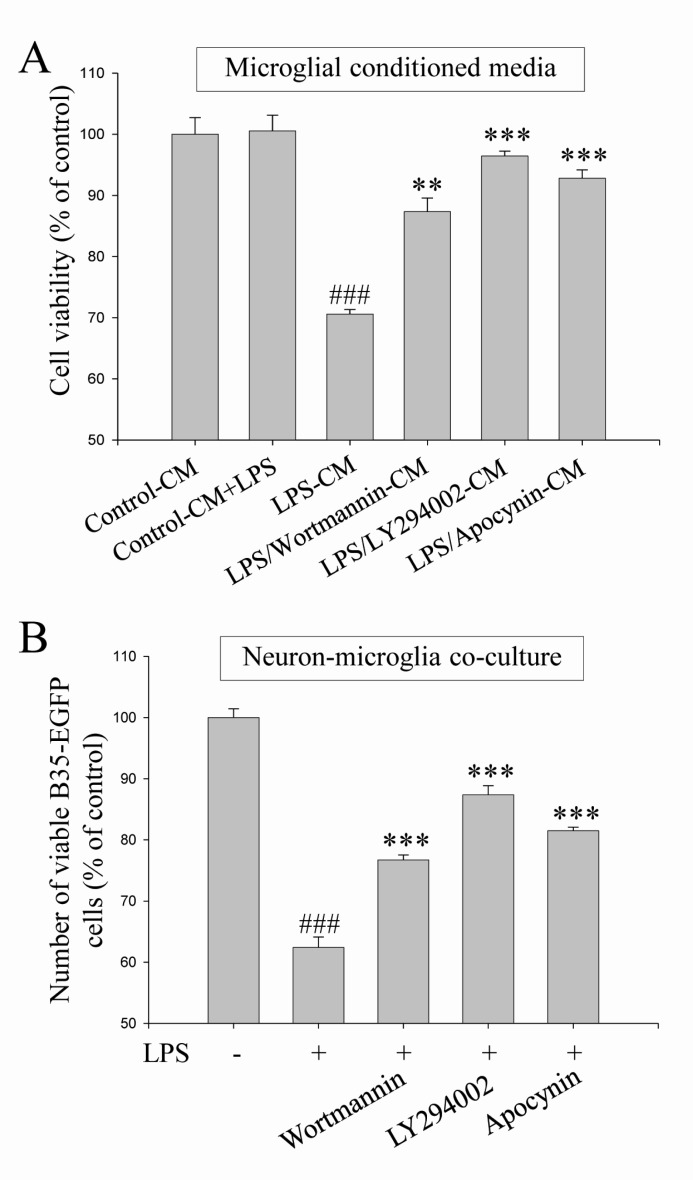
We demonstrated here that PI3K/Akt and NADPH oxidase-mediated pathways were suppressed by DEK treatment in activated microglia (Fig. 4 and 5). Therefore, neuroprotective effects of the blockade of PI3K/Akt and NADPH oxidase were investigated against microglia-mediated neuronal cell death in microglial conditioned media system and in neuron-microglia co-culture system. These two experimental models were also used to investigate the effect of DEK on microglia-mediated neuronal cell death (Fig. 2). Wortmannin (100 nM) and LY294002 (25 µM) as PI3K inhibitors and apocynin (500 µM) as a NADPH oxidase inhibitor were pretreated in both experimental systems and then, neuronal cell death was evaluated as shown in Fig. 2.
The results demonstrated that pretreatment of wortmannin, LY294002 and apocynin significantly attenuated microglia-mediated neuronal cell death in the microglial conditioned media system (Fig. 6A) and in the neuron-microglia co-culture system (Fig. 6B). Taken together, these results indicate that PI3K/Akt and NADPH oxidase-mediated signal pathways, which were suppressed by DEK, play a critical role in microglial inflammatory signaling and microglia-mediated neuronal cell death.
DISCUSSION
In the present study, we demonstrated, using both a microglia and neuron co-culture system and microglial conditioned media system, that DEK suppresses excessive microglial activation and subsequent neuronal cell death via downregulation of ERK, Akt, and NADPH oxidase-mediated pathways. These experimental systems have been successfully established in determining the neuroprotective activity of chemical compounds [3,21,22,23]. Since neurons were never exposed to DEK in this co-culture system and microglial conditioned media system, the neuroprotective effects of DEK are purely due to the suppression of microglia activation. Although this co-culture system is not the same as in vivo condition, it is believed to mimic the brain tissue where neurons and microglia mutually interact.
LPS is known to be a specific ligand for Toll-like receptor 4 (TLR4). Therefore, the LPS stimulation model is considered as a powerful experimental tool for TLR4-mediated microglial activation, which has been implicated in brain injury and neurodegeneration. It has been established that multiple signaling pathways are activated in microglial cells upon recognition of LPS by TLR4 [3]. DEK was previously shown to inhibit COX-2 and iNOS via p38 kinase and NF-κB, not ERK in activated microglia following LPS stimulation [14]. In spite of this report, the molecular mechanism underlying neuroprotective activity of DEK still remains to be elucidated. Therefore, we demonstrated here using a neuron-microglia co-culture system and microglial conditioned media system that DEK suppresses microglia-mediated neuronal cell death via suppression of microglial activation, which is mediated by downregulation of ERK, Akt, and gp91phox-mediated pathways.
A growing body of evidence has indicated that phosphorylation of MAPKs, Akt and IκB kinase (IKK) and NF-κB translocation were significantly suppressed by downregulation of intracellular ROS using ROS scavenger N-acetyl cysteine (NAC), suggesting these molecules are ROS-dependent in activated microglia [3,9,10,11,26,27]. The upstream and downstream relationships between these molecules have been proposed in several types of cells where NF-κB activity is associated with MAPKs or PI3K/Akt pathways. IκB degradation is known to be mediated through phosphorylation of IKK by Akt in activated microglia [28,29]. It is currently unclear whether MAPKs acts upstream of NF-κB in activated microglial cells, although NF-B activation via MAPKs pathway was demonstrated in other cell types [27,30].
In addition to these above pathways, we investigated the effect of DEK on NADPH oxidase-mediated pathway. We found here that blockade of NADPH oxidase using the well-known inhibitors DPI and apocynin significantly suppressed microglial pro-inflammatory mediator NO production (Fig. 5C), indicating that NADPH oxidase and NADPH oxidase-derived ROS are critical in the microglial activation pathway. We found that intracellular ROS generation was rapidly evoked within 5 min in LPS-stimulated microglia. Maximum induction was observed after 30 min and sustained until 24 h after LPS stimulation (Fig. 5E). LPS-induced intracellular ROS levels were significantly suppressed by DEK treatment in a dose-dependent manner (Fig. 5F), as consistent with the previous report [13].
On the other hand, the increases in gp91phox expression were significantly shown 6 h after LPS stimulation. The increased expression was maintained through 24 h (Fig. 5A). This gp91phox upregulation is likely to contribute to high levels of sustained intracellular ROS. Significant suppression of gp91phox expression could contribute to reduction of intracellular ROS levels in DEK-treated microglia, since gp91phox expression is the catalytic subunit of NADPH oxidase complex, the major source of microglial ROS production. Taken together, these results suggest that DEK suppresses NADPH oxidase-ROS pathway by not only scavenging intracellular ROS (Fig. 5D and F), but also suppressing gp91phox expression (Fig. 5B).
It is not clear how DEK suppresses gp91phox expression in LPS-stimulated microglia. However, it could be postulated that DEK might suppress gp91phox expression via down regulating the NF-κB pathway. The existence of typical NF-κB binding elements was demonstrated in the promoter of Nox1 and Nox4, which are also isoforms of catalytic subunits of NADPH oxidase complex. Transcriptional regulation of Nox1 and Nox4 by NF-κB was proposed in human aortic smooth muscle cells [31]. However, to our knowledge, transcriptional regulation of Nox2 by NF-κB has not been clearly elucidated yet.
The NADPH oxidase generates superoxide anion (·O2-) by reducing O2 in the extracellular space, which can be converted to other ROS molecules such as H2O2 and highly reactive hydroxyl radical (·OH) [32]. In addition, ·O2- toxicity is exacerbated by reacting with ·NO to form peroxinitrite (ONOO-), which modifies tyrosine in proteins to nitrotyrosines [33,34]. H2O2 is a diffusible molecule that can penetrate the lipid bilayer to enter the cytosol [32], where it can function as a second messenger for redox signaling [35]. It has been demonstrated that intracellular H2O2 inactivates protein tyrosine phosphatases (PTPs) by oxidizing the catalytic domain containing reactive cysteine residue, which leads to promote tyrosine phosphorylation of certain protein kinases in various cell types [35]. MAPKs phosphorylation, which is important in microglial activation signaling pathways, can be modulated by MAPKs phosphatases-1 (MKP-1) and apoptosis signal-regulating kinase-1 (ASK-1), which are known to be redox-regulated phosphatases and kinases respectively [36,37,38].
It is of interest that p38 activation was required for the phosphorylation of a cytosol subunit of NADPH oxidase complex p47phox and the subsequent ROS production in Mycobacterium tuberculosis-stimulated microglial cells [39]. In addition, NADPH oxidase-derived ROS was upstream signal molecules of p38, suggesting that neuroinflammation signals might be amplified through such mutual activation between MAPKs and NADPH oxidase-ROS pathway [38]. Similarly, there might be other positive feedback loop of mutual activation between NF-κB and NADPH oxidase-ROS pathway, considering that NF-κB regulates NADPH oxidase subunit expression via Nox1 or Nox4 transcriptional regulation [31] and, inversely, NADPH oxidase activates NF-κB through the subsequent ROS [3,9,11]. However, the existence of these possible multiple positive feedback loops in microglial activation process remains to be elucidated clearly.
Emerging evidence from several studies indicates that microglial TLR4 contributes to spinal nerve injury-induced neuropathic pain [40] and delayed neuronal cell death in a cerebral ischemia animal model [41]. These reports suggest that TLR4 signaling may play a pivotal role in the development of various neurodegenerative diseases. Previous reports have suggested that microglial TLRs may bind to not only exogenous LPS ligands but also various endogenous ligands released from damaged neurons or accumulated in certain pathological conditions [42,43]. Furthermore, we speculate that there might be crucial cross-talk in the downstream signaling pathways between TLR4 and certain microglial receptors for specific endogenous molecules such as ATP and glutamate [16], though these possibilities remain to be clearly elucidated.
In conclusion, we demonstrate evidences indicating that DEK suppresses LPS-induced excessive microglial activation and the subsequent neuronal cell death via downregulation of ERK, Akt, and NADPH oxidase-mediated pathways. It is postulated that the ROS scavenging activity of DEK seems to play a central role in the neuroprotective mechanism against microglia-mediated neurotoxicity, since these signaling pathways are known to be ROS-dependent [3,9,10,11,26,27]. Taken together, these results suggest that DEK might have a neuroprotective potential via attenuating microglia-mediated neurotoxicity implicated in the pathogenesis of neuroinflammation and neurodegeneration.
 XML Download
XML Download