

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The mammalian innate immune system acts like a guard and has ability to recognize microbial infection and plays its role in eradication of microbes. Inflammation is a primarily localized protective response by the host to eliminate destructive stimuli, and healing process for repairing injured tissue (1). Inflammation is characterized by five fundamental symptoms: redness, swelling, heat, pain, and loss of tissue function, which results in an increased permeability of the vascular endothelium as well as extravasations of immune cells (2). Macrophages and dendritic cells (DCs) are the central player in innate immunity; however, epithelial cells, endothelial cells, and fibroblasts also take part in innate immunity. Pathogen associated molecular patterns (PAMPs) are highly conserved among the microorganisms and germ line-encoded pattern recognition receptors (PRRs) are responsible for sensing of the microbes by these PAMPs (2, 3). Damaged cells releases endogenous molecules called as damage associated molecular patterns (DAMPs) which are also recognized by PRRs. The PRRs are divided into four different classes and called as Toll-like receptors (TLRs), C-type lectin receptors (CLRs), Retinoic acid inducible gene (RIG)-1-like receptors (RLRs) and NOD-like receptors (NLRs) (2~8).
Controlled inflammatory response is beneficial, as it provides protection against destructive stimuli while dysregulated inflammation is injurious because it results in different diseases such as rheumatoid arthritis, septic shock and cancer (1). Apart from the stimulus, inflammation is involved in an adaptive response for restoring homeostasis (1). Tissue macrophages and mast cells are involved in recognition of primary infection and lead to production of different inflammatory mediators, including pro-inflammatory cytokines and chemokines. The instantaneous effects of these mediators educe inflammatory exudates which contain plasma proteins and neutrophils (1).
In general, TLRs respond to extracellular stimuli while NLRs are involved in detection of cytosolic stimuli and provide crucial host defense against pathogens (5, 9). All TLRs recognize specific unique molecular patterns of microbes and elicit the innate immune system (10, 11). Myeloid differentiation primary response gene 88 (MyD88), toll-interleukin 1 receptor (TIR) domain containing adaptor protein (TIRAP)/MyD88 adaptor like (Mal), TIR domain-containing adapter-inducing interferon-β (TRIF), and TRIF-related adaptor molecule (TRAM) are TIR domain containing adaptors which are involved in regulation of TLR-mediated signaling pathways (12, 13). Most TLRs are involved in signaling by involving MyD88 with the exception of TLR3 which signals through TRIF. Deficiency of MyD88 leads to defects in production of IL-12 p40 and TNF-α, thus MyD88 plays an important role in regulation of innate immune response (12~14). The TRIF can also be used in combination with TRAM in the TLR4-MyD88-independent pathway (6, 15, 16). TLR3 employ TRIF to activate interferon regulated factor 3 (IRF3) which ultimately translocates to nucleus and results in expression of interferon (IFN)-β (15, 16).
Activation of TLRs results in recruitment of IL-1 receptor-associated kinases (IRAKs) family members through MyD88-dependent mechanism. The activated IRAK associates with downstream adaptor tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) and results in activation of TGF-β-activated kinase 1 (TAK1) (17). Activated TAK1 complex regulates IκB kinases (IKK) complex and mitogen activated protein kinases (MAPKs) which ultimately result in activation of NF-κB and AP-1, respectively (18). Finally activated NF-κB and AP-1 is responsible for production of proinflammatory cytokines including IL-12, IL-6 and TNF-α (19). The activation of NF-κB and MAPKs is tightly regulated by phosphorylation and ubiquitination and is responsible for production of TNF-α, IL-6 and IL-12 (20). Translocation of NF-κB to nucleus is also involved in expression of pro-forms of IL-1β and IL-18. Further processing and release of IL-1β and IL-18 is dependent on NLRs and caspase-1 (7, 21, 22). Recent reports have highlighted the roles of NF-κB and MAPK pathways in response to inflammatory conditions and infectious agents. Moreover, MAPKs also play critical role in regulation of inflammation-associated cancer development (23).
Mitogen-activated Protein Kinases
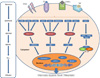
MAPKs were initially called as extracellular signal-regulated kinases (ERKs); a chain of proteins in the cell that is involved in communication of signals in eukaryotic cells. MAPK pathway plays critical role in communicating signals from receptors on the cell surface to the DNA in the nucleus to manage cellular functions such as proliferation, growth, differentiation, migration and death (24, 25) (Fig. 1). Binding of growth factor to the receptor on the cell surface results in initiation of signal transduction and activation of gene expression and then terminates this signal as negative feedback. Different MAPKs pathways have been characterized in mammals: ERK1 and 2, c-Jun amino-terminal kinases (JNK) 1, 2, and 3, p38 isoforms α, β, γ and δ, and ERK3 and 4, and ERK5 (26).
Various factors can activate MAPKs, but generally growth factors activate ERK1 and ERK2, whereas the JNK and p38 are activated in response to stress stimuli (27). All MAPKs have distinctive characteristics, but some features are common by the MAPK pathways and each family possesses conserved consecutively acting kinases; which includes MAPK, a MAPK kinase (MAPKK), and a MAPKK kinase (MAPKKK) (28). MAPKKK phosphorylates and activates a MAPKK, which ultimately activate MAPK through dual phosphorylation on threonine and tyrosine residues (28). MAPK not only mediates diverse range of functions by phosphorylation of phospholipases, transcription factors, and cytoskeletal proteins but also catalyzes the phosphorylation and activation of protein kinases, called MAPK-activated protein kinases (MKs) (28). The MKs are related kinases, involved in diverse range of biological functions and play their role as amplification step in MAPK catalytic cascades. The MK family consists of the mitogen- and stress-activated kinases (MSKs), the 90-kDa ribosomal S6 kinases (RSKs), the MAPK interacting kinases (MNKs), MK2, MK3, and MK5 (28). The activity of MAPKs is managed by phosphorylation of two residues; a threonine and a tyrosine, found in phosphorylation lip or activation loop (29, 30).
ERK MAPKs
The mammalian ERK1 and ERK2 (p44 and p42 MAPK) have more than 80% amino acid identity and copiously present in all tissues (27). Rapidly accelerated fibrosarcoma (Raf) kinases are a family of three serine/threonine-specific protein kinases, A-Raf, B-Raf, C-Raf, and play important role in the Ras-Raf-MEK-ERK signal transduction cascade (31). Rat sarcoma (Ras) is a guanosine-nucleotide-binding protein and function as binary signaling switches with "on" and "off" states (32). Stimulation of MAPKKK (A-Raf, B-Raf, Raf-1 and C-Mos) by mitogen directs the phosphorylation and activation of a MAPKK (MEK1 and MEK2) which then stimulates ERK1 and ERK2 activity through dual phosphorylation on tyrosine and threonine residues.
Stimulation of cell surface receptors such as tyrosine kinases and G protein-coupled receptors induce signal and activate Raf/MEK/ERK cascade (33). Activation of Raf leads to phosphorylation of MEK1 and MEK2 which ultimately phosphorylate ERK1 and ERK2 (34). ERK1/2 signaling plays an important role in regulation of cell proliferation. Activation of ERK1 and ERK2 leads to phosphorylation of different substrates which include some membrane proteins, cytoskeletal proteins, nuclear substrates and numerous MKs (26). In ear edema of mouse the activities of both ERK-1 and ERK-2 were upregulated and their phosphorylation was measurable after 15 minutes. Inhibition of MAPK/ERK pathway has been successfully used in treating inflammation in an ear edema of mice (35).
JNK MAPKs
JNKs are also called as stress-activated kinases (SAPKs), because initially they were characterized by their activation in response to cell stress (36, 37). Initially the first member of the JNK family was isolated from rat liver (38). JNK1, JNK2, and JNK3 are also called as SAPK γ, SAPK α, and SAPK β, respectively (27, 36). The JNKs can be activated by UV irradiation, growth factor deprivation, cytokines and DNA-damaging agents. All the JNKs are ubiquitously expressed; however, JNK3 is mainly expressed in brain, heart and testes (36, 39~42). Activation of MAPKKK (MEKK1-4, DLK, MLK2 Tpl-2, ASK1, TAK1 and TAO1/2) by stimuli results in activation of MAPKK, which includes MEK4 and MEK7 and are responsible for the activation of JNKs. The activation of JNK necessitates dual phosphorylation on tyrosine and threonine residues within a conserved Thr-Pro-Tyr (TPY) motif (36).
The transcription factor c-Jun is known as a substrate for JNKs. The JNKs are also involved in phosphorylation of other transcription factors which include activating transcription factor-2 (ATF-2), nuclear factor of activated T-cells cytoplasmic 1 (NF-ATc1), heat shock factor protein 1 (HSF-1), and Signal transducer and activator of transcription 3 (STAT3) (26, 36, 43). Roles of MK activated by JNKs still remain unclear. The activator protein 1 (AP-1) is a transcription factor which controls gene expression and can be activated by a variety of stimuli, including cytokines, stress, growth factors and infections (44). Phosphorylation of c-Jun, ATF2 and ATF3 substrates by JNKs results in enhancement of AP-1 controlled specific gene expressions (45). AP-1 is involved in managing several cellular processes such as proliferation, differentiation and apoptosis (46).
p38 MAPKs
Four p38 isoforms are found in mammals, which include: p38α, p38β, p38γ, and p38δ. Most of the tissues widely express p38α and p38β isoforms, whereas p38γ, and p38δ are expressed by kidney, skin and muscle cells (47). Defect in p38 was proposed to be involved in impaired vascularization of the placenta and results in embryonic death at the age of 10.5~16.5 in mice (48, 49). The p38 is also involved in insulin secretion in pancreatic β cells, as mice lacking p38 were found to be responsible for defects in insulin secretion (50). The p38 isoforms are strongly activated by stress and inflammatory cytokines in mammalian cells (51). The p38α gives response to heat shock and endotoxic lipopolysaccharide (47). The phosphorylation of p38 is managed by MKK3 and MKK6 which are under upstream control of MTK1 and ASK1 (28, 52). Activation of p38 results in phosphorylation of various cellular targets which include the microtubule-associated protein Tau, cytosolic phospholipase A2, and the transcription factors ATF1, ATF2, MEF2A, Sap-1, Elk-1, NF-κB, Ets-1, and p53. The p38 is also involved in activation of some MKs which include MSK1 and MSK2, MNK1 and MSK2 (36).
Conclusion and Perspective
Roles of TLRs and MAPKs in inflammation have been well characterized. All TLRs recognize distinctive PAMPs by involving some intracellular adaptors and MAPKs lead to expression of response to stimuli. MAPK signaling cascades are involved in regulation of key cellular functions which include cell proliferation, gene expression, cell motility and cell survival or death. A wide range of inhibitors of MAPK signaling pathways are available and scientists are working on identifying more natural MAPK inhibitors which will be helpful in treating inflammatory conditions and cancer.
 XML Download
XML Download