

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Regulatory T (Treg) cells are a distinct lymphocyte lineage endowed with inhibitory properties that affect the activation of the immune system. Treg cells (also called suppressor T cells) were first described in the early 1970s in a series of papers concerning thymic suppressor cells that could inhibit cell-mediated immunity in many different in vivo models (1-7) and that were characterized by the expression of CD25 and the Treg-specific forkhead/winged helix transcription factor 3 (Foxp3), which is required for their development and function (8-12). These cells can inhibit activation of other T cells and are needed for protection against autoimmune diseases and prevent rejection of allogeneic transplants (6,7,13-15). In humans, mutations in Foxp3 result in an autoimmune syndrome termed IPEX (immunodysregulation, polyendocrinopathy enteropathy, X-linked syndrome), an X-linked immunodeficiency syndrome characterized by insulin-dependent diabetes, thyroiditis, massive T cell infiltration in multiple organs, and chronic wasting (8,9,16). However, immunoregulatory function of Treg cells may hinder the induction of immune responses against cancer and infectious agents (14,17,18). Counteracting Treg cell activity can evoke effective antitumor immunity (18-24), and inhibition of Treg cell function in patients with cancer is an essential step to improve the efficacy of antitumor therapies, especially those based in immunotherapeutic approaches (11,16,18,21,23-30).
MicroRNAs (miRNAs) are small (19-22-nt) noncoding RNA molecules that are single-stranded in the functional form and derived from hairpin-structured precursors (31). Unlike their small size, they play an important role in the regulation of gene expression at the post-transcriptional level. After their discovery in Caenorhabditis elegans (32-34), there have been a large number of studies identifying miRNA in animals, plants, and viruses. Since the initial observation, more than 700 miRNAs have been identified in mammalian cells and have been shown to play important roles in human development, cellular differentiation and homeostasis, adaptation to the environment, oncogenesis, and host cell interactions with pathogens (35-41). Since miRNAs act as key regulators in a wide variety of biological processes, it is now apparent that abnormal miRNA expression is a common feature of various diseases (42-54). Recently, more miRNAs have been reported to be involved in the regulation of immune systems, demonstrating that miRNAs modulate many aspects of the immune responses such as differentiation, proliferation, cell fate determination, function of immune cells, and cytokine responses, as well as the intracellular signaling pathways (55-69). They function by directly binding to the 3' untranslated regions (3'UTRs) of specific target mRNA, leading to the repression of protein expression and the production of target mRNA degradation. So far, over 700 different miRNAs have been identified in the human genome (35,36). Each miRNA could have the potential to repress the expression of many, perhaps hundreds, of target genes. More than 100 different miRNAs are expressed by cells of the immune system and they have the potential to broadly influence the molecular pathways that control the development and function of innate and adaptive immune responses (35,57,58,65-67,70,71). The expression of miRNA is also markedly dysregulated in cancers of immunological origin, in which they are thought to have tumor-suppressive or tumor-promoting activities depending on the nature of their specific target mRNA (35,49,72-83). Now, miRNA studies are resolving some unsolved issues in immunology. Recent studies have shown that miRNAs have unique expression profiles in cells of the innate and adaptive immune systems, and have pivotal roles in the regulation of both cell development and function (18,35,55,67,71,84-88). Furthermore, when miRNAs are aberrantly expressed they can contribute to pathological conditions involving the immune system, such as cancer and autoimmunity (35,43,68,89). They have also been shown to be useful as diagnostic and prognostic indicators of disease type and severity (35,82,90-95).
It is, therefore, not surprising that these novel class of cellular regulators have been shown to be involved in regulation of various processes for cell fate determination, such as cell cycle, proliferation, apoptosis, differentiation and development of nervous, skeletal and cardiac muscle tissues and haematopoietic stem cells (96,97). The de-regulation of such endogenous and epigenetic transcriptional networks involving miRNA may be relevant to disease pathogenesis (43,89,96,98,99). Recent studies have also implicated aberrant expression of specific miRNAs in a broad spectrum of human diseases, including diabetes (50,54), cardiovascular disease (47,100) cancer (42,72-79,83,101-104), endotoxin shock (105), neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, spinocerebellar ataxia, Huntington's disease (45,106), psychiatric disease, such as schizophrenia, bipolar disorder and autism (43), rheumatic diseases (89,107-109), ethanol addiction (48), renal disease (50), endometriosis (51), asthma (52), and prion disease (53).
The first indication that miRNA might regulate the immune responses was a report in 2004 showing selective expression of miR-142a, miR-181a and mir-223 in immune cells (38). In this study, miR-181a was localized in B-lymphocytes, miR-142a in B-lymphocytes and myeloid cells whilst miR-223 was confined to myeloid cells (38). Since this initial observation, miRNAs have been implicated in the regulation of maturation, proliferation, differentiation, and activation of immune cells (65).
Treg CELLS
CD4+ T cells orchestrate diverse modes of immune responses by differentiating into distinct helper subsets, characterized by unique patterns of cytokine secretion (110). There are three subsets: Th1 cells, which secrete the signature cytokine interferon-gamma (IFN-γ), Th2 cells, which secrete the signature cytokine interleukin (IL)-4 and Th17 cells, which secrete IL-17 (110). Dysregulated differentiation of CD4+ T cells can lead to defects in the development of pathogen-specific immune response and can also result in lymphocyte-mediated disease (110).
There are at least three recognized subsets of CD4+ Treg cells involved in the negative regulation of immune response (16,111-115). Type 1 Treg (Tγ 1) cells are induced in the periphery and suppress T cell proliferation through the production of IL-10 and transforming growth factor-β (TGF-β), and do not have a unique cell marker, but are identified by their production of IL-10 and not pro-inflammatory cytokines. T helper 3 (Th3) cells are a regulatory T cell population that originates in the periphery and mediates suppression through the secretion of TGF-β. Importantly, CD4+CD25+Foxp3+ Treg cells are classical Treg cells. Depletion of these cells has attracted much attention in recent years (11,16,22,23,28,116,117). CD4+CD25+Foxp3+Treg cells, according to their sources, can be divided into two classes: naturally occurring Treg (nTreg) cells and adaptive or inducible Treg (iTreg) cells. nTreg cells are generated in the thymus during development of T cells (112,118). These cells, which constitute 5~10% of the total lymphocytes, enter the peripheral circulation and are widely distributed in peripheral reservoir lymph nodes and spleen (12,112,113,115,117). Thymus-derived CD4+ CD25+Foxp3+Treg cells are stable with respect to retaining regulatory function and Foxp3 expression in the periphery (117). On the contrary, iTreg cells are developed in the periphery from naive T cells or nTreg cells under the influence of various inductive signals, most importantly TGF-β or IL-10 (27,111,112,119). Treg cells are primarily characterized by CD4+Foxp3+ or CD4+CD25+Foxp3+ T cells, and Foxp3 has been considered as a master regulatory transcription factor for Treg cells (10-12,15,116). Recently, Taylor and Llewelyn reported that superantigen (staphylococcal enterotoxin A or streptococcal pyrogenic exotoxin A) can induce Treg cells in human peripheral blood mononuclear cells (PBMCs) and approximately 2% of circulating T cells are nTreg cells (120). Thornton et al. (121) reported that in both mouse and human, approximately 70% of peripheral Foxp3+ Treg cells express the transcription factor Helios. Interestingly, they found that Foxp3 cells generated in the thymus all co-expressed Helios, whereas Treg cells generated in vitro from conventional peripheral T cells with anti-CD3 and anti-CD28 antibody stimulation in the presence of IL-2 and TGF-β1 do not express Helios. This, they suggested, could mean that approximately 70% of Foxp3+ Treg cells in the periphery are thymically-generated, and 30% are derived from peripheral conversion of conventional T cells. These results demonstrate that Helios is potentially a specific marker of thymic-derived Treg cells and raises the possibility that a significant percentage of Foxp3+ Treg cells are generated extrathymically. It has also been reported that Foxp3+ Treg cells also generated outside the thymus under a variety of conditions (12,27,122).
Very currently, various research groups have reported many interesting findings. Kerdiles et al. reported that T cell-specific loss of Foxo1 resulted in exocrine pancreatitis, hind limb paralysis, multi-organ lymphocyte infiltration, anti-nuclear antibodies, and expanded germinal centers, and that Foxo transcription factors are essential for the development and function of Foxp3-expressing Treg cells by controlling the expression of genes associated with Treg cell function (123). Procaccini et al. reported that the anergic state of Treg cells depends on the elevated activity of the mammalian target of rapamycin (mTOR) pathway induced by leptin: transient inhibition of mTOR with rapamycin, before T cell receptor stimulation, made Treg cells highly proliferative in the absence of exogenous IL-2 (124). Treg cells from aged mice are more resistant to apoptosis than Treg cells from young mice and, notably, aged Treg cells exhibit decreased expression of the proapoptotic molecule Bim compared with Treg cells from young mice (125). Casares et al. reported that a 15-mer synthetic peptide designated P60 enters the cells, inhibits Foxp3 nuclear translocation, and reduces its ability to suppress the transcription factors nuclear factor-kappa B (NF-kB) and nuclear factor of activated T-cells (NFAT) (29). They also reported that P60 induces protection against tumor implantation and, similarly, improves the antiviral efficacy of a recombinant adenovirus expressing NS3 protein from hepatitis C virus. The data indicate that functional inhibition of Treg cells by the Foxp3-inhibitory peptide P60 may enhance antitumor and antiviral immunotherapies (29).
Interestingly, systemically applied glucocorticosteroids (GCSs) endow epidermal Langerhans cells with Treg-promoting properties, which have shed new light on the mechanisms of GCS-mediated immunosuppression (126). Eller et al. reported that IL-9 production by Treg cells recruits mast cells, providing the first direct in vivo evidence that the nephroprotective, anti-inflammatory effects of Treg cells critically depend on IL-9-mediated attraction of mast cells into kidney-draining lymph nodes (127). Interestingly, the vitamin D analog, TX527, promotes a human CD4+CD25highCD127low Treg cell profile (128). T-cell activation and function require a structural engagement of antigen-presenting cells and these cell contacts are characterized by two distinct dynamics in vivo: transient contacts resulting from promigratory junctions called immunological kinapses or prolonged contacts from stable junctions called immunological synapses (IS). Comparison of the ability of Treg cells and Th cells to form IS on supported planar bilayers revealed that human Treg cells form more stable IS than do Th (129). Synapse stabilization may enhance Treg function, as suggested by the recent evidence that IS play an important role in Treg effects mediated through dendritic cells (DCs) (129).
Humans harbor nearly 100 trillion intestinal bacteria that are essential for health. Millions of years of co-evolution have molded this human-microorganism interplay into a symbiotic relationship, in which gut bacteria make essential contributions to human nutrient metabolism and in return occupy a nutrient-rich environment (110).
There is also evidence that Treg cells are induced by intestinal bacteria or their molecular products, such as the polysaccharide A carbohydrates expressed by Bacillus fragilis and the non-culturable Clostridia-related segmented filamentous bacteria, and that there is spontaneous intestinal inflammation in mice with specific deficiencies in regulatory cytokines (such as IL-10 and TGF-β) or in factors that determine Treg cell thymic selection (such as autophagy-related gene; 5), differentiation and/or function (such as Foxp3 or αVβ8 integrin) (110). These findings clearly indicate that Treg cells play an important role in immune adaptations that maintain intestinal homeostasis with intestinal microorganisms.
miRNA BIOGENESIS
Small RNAs can be classified into different groups based on their origin or the components to which they coupled (31,39,130,131). These small RNA groups include miRNAs, small-interfering RNAs (siRNAs), trans-acting siRNAs (tasiRNAs), small-scan RNAs (scnRNAs), repeat-associated siRNAs (rasiRNAs), and Piwi-interating RNAs (piRNAs). siRNAs are the products of long, Dicer-processed, double-stranded (ds) RNAs that silence genes by cleaving their target mRNAs (31,36,130). Regardless of the type and size, small non-coding RNAs share one unifying function in cellular physiology - epigenetic regulation of gene expression (35,37).
The process of miRNA biogenesis and target mRNA repression have been extensively studied and reviewed (31,37,48,55,96,132). Human miRNA are present in introns of coding genes and introns and exons of noncoding transcripts. To generate mature miRNA, primary miRNAs (pri-miRNAs) from long primary transcripts go through a series of endonucleolytic steps. Pri-miRNAs containing a 5'7-methylguanosine cap and 3' poly (A) tail are transcribed by RNA polymerase II, whereas others are transcribed by RNA polymerase III (130,133). The pri-RNAs are then recognized and cleaved by the Drosha-DGCR8 (DiGeorge syndrome critical region 8 homologue) microprocessor complex to yield an approximately 70nt intermediate with the typical stem-loop hairpin structure, precursor miRNAs (pre-miRNAs), in the nucleus (134,135). After transported to the cytoplasm by the RanGTP-dependent dsRNA-binding protein Exportin 5 (135), the pre-miRNAs are further processed into an approximately 22nt double-stranded miRNA duplex by the cytoplasmic RNAase III enzyme Dicer (136). One strand of this miRNA duplex, which is designated as the guide strand, is incorporated into a large protein complex, miRNA-induced silencing complex (miRISC), formed by Dicer, TRBP (human immunodeficiency virus 1-transactivating response RNA-binding protein, a dsRNA-binding domain protein) and Ago2 (Argonaute protein-2), to yield the mature miRNA. miRISC is then transported by importin 8 to its cognate mRNA, leading to repression of the target. Finally, miRNA can be localized to specific organelles, such as stress granule or processing (P-) bodies, some of which are thought to be used during stress (35). At the same time, the other strand, designated the passenger strand, is degraded (Fig. 1).
Each mature miRNA interacts with a specific mRNA, typically through pairing of nucleotide bases between the miRNA sequence and complementary sequences in the mRNA 3'UTR. As gene regulators, the functions of miRNA are mediated through translational repression or mRNA degradation. Generally, whether target mRNAs are cleaved and degraded is mainly determined by the complementarity between miRNAs and target mRNAs (31,40,41,55). miRNAs are believed to either repress mRNA translation or reduce mRNA stability following imperfect binding between the miRNA and the miRNA-recognition elements (MRE) within the 3'UTR of target genes (31,41,137). Interestingly, miRNAs display a saptiotemporal pattern of expression and function and, therefore, are crucial in the regulation of various biological processes in various stage (35,37,41).
miRNAs IN THE INNATE IMMUNE RESPONSE
miRNAs play a key role in diverse biological processes, such as embryogenesis, differentiation, inflammation, viral infections, and carcinogenesis (58). The importance of miRNAs on immune system development and response has been reported, as has the involvement of miRNAs in the regulation of immunity, including the development and differentiation of immune cells, antibody production, and inflammatory mediator release (137). The innate immune system is characterized by rapid response to pathogens and is mediated by macrophages, granulocytes, DCs and natural killer (NK) cells (138,139). In contrast, the adaptive immune system, which is composed of T and B cells, is characterized by memory or secondary antigen-specific immune response. The NK T-cell system bridges innate and adaptive immunity (139).
miRNA-mediated gene regulation is critical for normal cellular functions, and as much as one-third of human mRNAs may be miRNA targets (140). Emerging evidence suggests that miRNAs play a key role in the regulation of immunological functions including innate and adaptive immune responses, development, and differentiation of immune cells, and the prevention of autoimmunity (55-58,140) (Fig. 2). Mice deficient for bic/miR-155 are immunodeficient and bic/miR-155 is required for the function of B-cells, T-cells, and DCs (59), indicating that bic/miR-155 plays a key role in the homeostasis and function of the immune system. The evolutionarily conserved miR-155 has an important role in the mammalian immune system, specifically in regulating T helper cell differentiation and the germinal center reaction to produce an optimal T-cell dependent antibody response (71). Innate immunity is a physiogenetically ancient biological system that multicellular organisms have evolved to defend themselves from invading pathogens. The job of recognition of the "nonself" from "self" in this system belongs to pattern recognition receptors (PRRs). Acting as molecular sentinels, PRRs detect highly conserved microbial molecules (pathogen-associated molecular patterns; PAMPS) and elicit pathogen-specific cellular responses that result in elimination of intruders as well as, in higher vertebrates, mobilization of the adaptive immune system (61). There is evidence that miRNAs of the host may impinge on the viral life cycle, viral tropism, and the pathogenesis of viral diseases (64). For example, human miR-32 has a direct negative effect on the replication of retrovirus primate foamy virus type 1 (61,64).
To elucidate the role of miRNA in the immune response in vivo, Moschos et al. (141) examined the different expression of 104 miRNAs using real-time polymerase chain reaction (PCR) during the innate immune response in the mouse lung following exposure to aerosolized lipopolysaccharide (LPS). Following challenge, rapid and transient increase in both the mean (4.3-fold) and individual levels of miRNA expression occurred and peaked at 33 h. Crucially, this increase was correlated with a reduction in the expression of TNF-α, keratinocye-derived chemokine, and macrophage inflammatory protein-2, suggesting a potential role for miRNAs in the regulation of inflammatory cytokine production. Examination of the individual miRNA expression profiles revealed a time dependent increase in miR-21, -25, -27b, -100, -140, -142-3p, -181c, -187, -194, -214, -223, and -224 (141).
Recent functional analysis of miRNAs in the immune system revealed that many cellular decisions are controlled by single miRNAs that entail significant down-regulation of one of few target proteins (71). Investigation of miRNA/mRNA pairs showed that miRNA-adjusted target protein levels are crucial at specific cellular transition points (71).
While the role of miRNAs in cell fate decisions linked to proliferation, differentiation, and apoptosis has long been recognized, the importance of these noncoding small RNAs on immune system development and response has only recently become evident. In addition to facilitating cell fate decisions of immune cells (e.g. miR-181a and miR-223), miRNAs also regulate central elements of the adaptive immune response such as antigen presentation (e.g., miR155) and T-cell receptor signaling (e.g., miR-181a) (63). Furthermore, the same study reported that miRNAs are involved in innate immunity through regulation of TLR signaling and cytokine response (e.g., miR-146). Intriguingly, cellular miRNAs not only alter immune cell development and function, but are also able to directly affect viral replication (63). Conversely, virus-encoded miRNAs shape the host-virus interactions and regulate the viral life cycle (63,64,142).
In contrast to miR-146, which is mainly regulated by bacterial products, miR-155 is also regulated by virally relevant stimuli such as the synthetic TLR3 ligand poly(I:C) (64). In addition to TLR-mediated sensing of bacterial and viral pathogens, miR-155 expression is also strongly induced by inflammatory cytokines such as antiviral cytokines IFN-β, IFN-γ through TNF-α autocrine/paracrine signaling, suggesting that miR155 is a component of the innate immune response to a broad range of inflammatory mediators (64). As discussed above, miR-155 is also essential for proper T-cell and B-cell response, as well as for interaction between DCs and T-cells. In contrast to miR-155, miR-146, and miR-137, another miRNA, miR-125b, is suppressed by LPS-treatment in a murine macrophage cell line (105). The function of miR-125b in macrophages may be to ensure that the LPS pathway is turned off in the absence of microbial infection, while its down-regulation in response to LPS may be needed for the macrophage inflammatory response (105). It seems clear that miRNAs are involved in the regulation of innate immune response.
Cytokine stimulation of immune cells also leads to a rapid shift in miRNA levels. For example, miR-125b and miR-155 levels oscillate within 1 h of TNF stimulation in mouse Raw 264.7 cells (105). These changes in miRNA levels enable the immune cells to limit the level and duration of immune responses to infections so that these responses do not become detrimental to the health of the organisms. Rapid changes in the levels of several miRNAs, however, were reported in the Huh7 hepatoma cells ltreated with IFN-β (63).
Recent studies indicate that those miRNAs that are selectively and/or highly expressed in immune cells, including the miR-17-92 cluster, miR-150, miR-155, miR-181, and miR-223, have a 'permissive' function in the maturation, proliferation, and differentiation of myeloid and lymphoid cells (65). Importantly, these actions of miRNAs often involve interactions with transcription factors. In contrast, the rapid and transient induction of miR-9, miR-146a, and miR-155 may negatively regulate the acute responses following activation of innate immune response through the down-regulation of proteins involved in the receptor-induced signaling pathway (65).
Acute retroviral infection provokes rapid and striking innate immune responses in what has been termed a cytokine storm. These response are crucial in determining the course of disease, as a delicate balance must be achieved between pro-inflammatory and anti-inflammatory processes. The former must be sufficiently ferocious to dampen viral replication and impede further infection, and the latter is needed to prevent the tissue damage inherent in chronic activation of the immune system (64,67,143). Human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV) infection of macrophages induces IFN-β, with both proinflammatory and anti-inflammatory roles, which is the main type 1 IFN induced during the initial innate response to retroviral infections of the central nervous system (CNS) (67). Interestingly, Witwer et al. recently reported that miRNAs, including miR-26a, -34a, -145, and let-7b, may directly regulate IFN-β in human and macaque cells (67). In primary primate macrophages, the main cell type implicated in HIV and SIV infection in the CNS, specific miRNAs are reduce, whereas miRNA inhibitors enhance, IFN-β protein production. These findings may have significant implications for the fine-tuning of innate immune response during retroviral infection and, potentially, for the therapeutic modulation of innate immune responses (67).
Interestingly, Kosaka et al. profiled miRNA expression in human breast milk and detected high expression levels of immune related miRNAs in the first 6 months of lactation (62). Using a miRNA microarray, they detected 282 of 723 known human miRNAs. More interestingly, several immune related miRNAs were abundant in milk: miR-155, a regulator of T- and B-cell maturation and the innate immune response; miR-181a and miR-181b, which are regulators of B-cell differentiation and CD4+ T-cell selection; the miR-17 and miR-92 cluster, a ubiquitous regulator of B-cells, T-cells, and monocyte development; miR-125b, a negative regulator of TNF-α production, activation, and sensitivity; miR-146b, a negative regulator of the innate immune response; miR-223, a regulator of neutrophil proliferation and activation; and let-7i, a regulator of TLR4 expression in human cholangiocytes (62). In contrast, T- and B-cell regulatory miR-150 was not detected. In addition, it has been reported that other body fluid and serum contain sufficiently stable miRNA signature (95,144-148). Together, these findings suggest that human breast milk contains miRNAs capable of transfer to immune cells to support the development of an infant's immune system and that human-to-human transfer of genetic material as miRNA occurs by means other than through sexual reproduction (62,95).
miRNAs IN MACROPHAGES/MONOCYTES
LPS stimulation up-regulates or represses the expression of miRNAs in macrophages. Among the LPS-responsive miRNAs characterized to date, miR-155 is up-regulated in response to LPS (105,149), promoting the expression of TNF-α and destabilization of PU.1, a key mediator of monocyte and macrophage differentiation, and TLR4 transcription (150). miR-155 also targets and suppresses SOCS1 (suppressor of cytokine signaling 1), a critical regulator of STAT (Signal Transducer and Activator of Transcription) signaling in Treg cells (151) and inositol phosphatase SHIP1 (35,55,149). In contrast, miR-125 inhibits the expression of TNF-α and is down-regulated in LPS-treated macrophages (105).
In the last decade, members of the Toll-like receptor (TLR) family have emerged as the primary evolutionarily conserved sensors of PAMP (64,152). Innate immune response to microbial infections occurs primarily through TLRs, which recognize PAMP and elicit the biosynthesis of proinflammatory cytokines from macrophages and other cells (152). MAPK (mitogen-activated protein kinase) phosphatase (MKP-1) is an archetypical member of the dual-specificity phosphatase family that deactivates MAPKs. Zhu et al. showed that MKP-1 is a direct target of miR-101 (a tumor-related miRNA) and that expression of miR-101 is induced by multiple TLR ligands, including LPS or peptidoglycan (152). Moreover, they also showed that treatment of cells with dexamethasone, a widely used anti-inflammatory agent, markedly inhibits miR-101 expression and enhances the expression of MKP-1 in LPS-stimulated macrophages. Together, these data indicate that miR-101 regulates the innate immune responses of macrophages to LPS through the targeting of MKP-1.
To examine the potential involvement of miRNAs in the regulation of the innate immune response, Taganov et al. analyzed the expression of 200 miRNAs after exposure of THP-1 cells to LPS. They found that production of mature forms for several of them (miR-146a/b, miR-132, and miR-155) is induced and that analysis of miR-146a and miR-146b gene expression revealed a pattern of induction in response to a variety of microbial components and proinflammatory cytokines (153). Additionally, they determined that TNF receptor-associated factor 6 (TRAF-6) and IL-1 receptor associated kinase (IRAK) represent potential molecular targets of miR-146 through an experiment with 3' UTR luciferase reporters. These findings suggest that miR-146a may function as a novel negative regulator that helps to fine-tune the immune response (153).
MiR-155 is one of the first miRNAs linked with cancer. To test whether virally relevant stimuli induce expression of specific miRNAs, O'Connell et al. used microarray technology to identify miRNAs induced in primary murine macrophages after exposure to polyriboinosinic:polyribocytidylic acid [poly(I:C)] or IFN-β (149). It was found that miR-155 was the only miRNA of those tested that was substantially up-regulated by both stimuli. Further investigation revealed that several TLR ligands increased miR-155 expression through either MyD88 (myeloid differentiation factor 88) or TRIF (TLR domain-containing adaptor-inducing IFN-β) signaling, whereas IFNs required TNF-α autocrine signaling to up-regulate miR-155. Inhibition of c-Jun N-terminal kinase (JNK) blocked both poly(I:C) and TNF-α induction of miR-155, indicating a role for MAPK signaling in the regulation of miR-155 levels (149). Together, because miR-155 is known to be function as an oncogene, these results reveal miR-155 as a component of the inflammatory response and suggest that this oncogenic miRNA may prove to be a link between inflammation and cancer. Tili et al. reported that miR-155 and miR-125b play important roles in the innate immune response (105). They also found that LPS stimulation of mouse macrophages resulted in the up-regulation of miR-155 and down-regulation of miR-125 levels. Furthermore, levels of miR-155 and miR-125b in macrophages displayed oscillatory changes in response to TNF-α. They also showed that miR-155 most probably directly targets transcript coding for several proteins involved in LPS signaling, such as the Fas-associated death domain protein (FADD), IκB kinase ε (IKKε), and the TNF receptor (TNFR superfamily)-interacting serine-threonine kinase 1 (Ripk1), while enhancing TNF-α translocation. Additionally, they found that Eµ-miR-155 transgenic mice produced higher levels of TNF-α when exposed to LPS and were hypersensitive to LPS/D-galactosamine-induced septic shock (105). These results may suggest the positive role of miR-155 in the regulation of the release of inflammatory mediators during the innate immune response. Interestingly, in miR-155 knock-out mice, miR-155 was also verified as a requirement for normal immune function (59). Rodriguez et al. have shown that bic/miR-155 regulates the function of T- and B-lymphocytes and DCs, leading to an overall diminution of immune responses (59). Identification of multiple novel potential targets of miR-155 supports the view that bic/miR-155 is a core regulator of gene expression in multiple cell types, with a "targetome" optimized to modulate the immune response (59). Moschos et al. investigated miRNA expression in mouse lungs after LPS exposure and found a rapid and transient increase in the expression of miRNAs. Particularly, 12 miRNAs (miR-21, miR-25, miR-27b, miR-100, miR-140, miR-1423p, miR-18c, miR-187, miR-194, miR-214, and miR-223) were regulated in a time-dependent manner (141). Of note, miR-146 and miR-155 were not up-regulated, in contrast to results obtained in macrophages. Interestingly, the lack of effect of dexamehasone in either control or challenged animals implies that the actions of glucorcorticoids per se are not mediated through changes in miRNA expression and that LPS-induced increases in miRNA expression are not mediated via classical inflammatory transcription factors.
Interestingly, Sonkoly et al. recently found miR-146 to be associated with psoriasis, a chronic inflammatory skin disease, indicating that alteration in the fine-tuning of the innate immune response by miRNAs may contribute to inflammatory disorders (64). Effective recognition of viral infection and subsequent triggering of antiviral innate and adaptive immune response are essential for the survival of the host. Upon recognition of viral components, host cells are activated to produce type I IFN and pro-inflammatory cytokines (64). Viruses are recognized by PRRs, including TLRs and retinoic acid-inducible gene 1 (RIG-1)-like helicase (RLH). RIG-1 was identified as a cytoplasmic viral RNA detector (149). Hou et al. analyzed the expression profile of miRNAs in mouse peritoneal macrophages during vesicular stomatitis virus (VSV) infection (154). They found that several miRNAs, including miR-146a, miR-155, miR-7a, miR-574-5p, and miR-125a-5p were significantly up-regulated and that VSV infection could up-regulate miR-146a expression in macrophages through a MyD88-independent, but RIG-1-NF-κB-dependent, manner. Importantly, they also demonstrated that miR-146a negatively-regulated VSV infection-triggered type I IFN production in a feedback reaction by impairing the RIG-1 pathway through the targeting of TRAF6, IRAK1, and IRAK2. Furthermore, they discerned associations of IRAK1 and IRAK2 with FADD (Fas-associated death domain), an important adaptor in RIG-1 signaling, in a VSV infection-inducible manner. The collective data from their study demonstrated that miR-146a is a feedback negative regulator of the RIG-1 signaling and RIG-1-dependent anti-viral pathways (154).
Activation of macrophages by LPS initiates feedback inhibitory loops that aim to establish tolerance to a subsequent LPS stimulus. Negative feedback signals either inhibit the expression of genes that are required for the response to LPS, such as the LPS receptor TLR4, or promote the expression of genes that inhibit the response, such as SOCS1 and IRAK-M. Negative regulators of TLR signaling such as SOCS1 and IRAK-M are up-regulated within 2 h from the start of the stimulation (105,151,153). Akt (also known as PKB) is a family of serine-threonine protein kinases that are activated by PI-3K (phosphoinositide-3 kinase) and that play key regulatory roles in a host of cellular functions including cell survival, cell proliferation, differentiation, and intermediary metabolism (155). Stimulation of macrophages with the TLR4 agonist LPS activates PI-3K and its downstream targets, including Akt kinase. Thus, activation of the PI-3K-Akt pathway suppresses LPS-activated MAPK and NF-κB signaling cascades in monocytes and DCs, resulting in decreased production of TNF-α and other cytokines (155). Interestingly, Androulidaki et al. investigated the role of Akt1 in the regulation of miRNA expression in LPS-stimulated macrophages (155). They found that LPS-treated Akt-/- macrophages failed to induce miRNA let-7e and expressed higher amounts of miR-155, in contrast to Akt+/+ macrophages. In silico analysis and transfection studies revealed that let-7e controlled TLR4 and miR-155 repressed SOCS1, and that these miRNAs were under the control of Akt1. As a result, Akt-/- macrophages were hyper-responsive to LPS and failed to develop endotoxin tolerance in culture and in vivo. In addition, they showed that introduction of these miRNAs in Akt1-/- macrophages restored their response to LPS (155). These results indicate that Akt1 controls macrophage responsiveness to LPS endotoxin tolerance by regulating the expression of miRNAs.
miRNAs IN GRANULOCYTES
Hematopoiesis is a life-long, highly-regulated multistage process wherein a pluripotent self-renewing hematopoietic stem cell (HSC) gives rise to all blood cell lineages. A recent study showed that hematopoietic lineage differentiation, the process of continuous development of HSCs into at least eight different blood lineage, is controlled by miRNAs (38). As mentioned earlier, three miRNAs that are preferentially expressed in murine hematopoietic tissues have been discovered: miR-181, miR-142, and miR-223 (38). miR-181 is very strongly expressed in the thymus, brain, and lung, and miR-223 is nearly exclusively expressed in bone marrow, whereas miR-142 is expressed in all hematopoietic tissues (38). Interestingly, the ectopic expression of miR-181 in mouse hematopoietic precursor cells leads to an increased fraction of B lineage cells (38).
Fazi et al. showed that human granulocytic differentiation is controlled by a regulatory circuitry involving miR-223 and two transcriptional factors, NF-1A (nuclear factor-1A) and C/EBPα (the CCAAT enhancer proteins). These two factors compete for binding to the miR-223 promoter. NF-1A maintains miR-223 at low levels, whereas its replacement by C/EBPα, following retinoic acid (RA)-induced differetiation, up-regulates miR223 expression (156). The data from the study indicated that miR-223 plays a crucial role during granulopoiesis and point to NF1-A repression as an important molecular pathway mediating gene re-programming in this cell lineage (156).
Neutrophils are an essential part of the innate immune response as they are critical for the first line of defense against bacteria and fungi. Using a loss-of-function allele in mice, Johnnidis et al. reported that the myeloid-specific miR-223 negatively regulates progenitor proliferation and granulocyte differentiation and activation, and that miR-223 mutant mice have an expanded granulocytic compartment resulting from a cell-autonomous increase in the number of granulocyte progenitors (157). They also demonstrated that Mef (myeloid ELF-1-like factor) 2c, a transcription factor that promotes myeloid progenitor proliferation, is a target of miR-223, and that genetic ablation of Mef2c suppresses progenitor expansion and corrects the neutrophilic phenotype in miR-223 null mice. Additionally, granulocytes lacking miR-223 are hypermature, hypersensitive to activating stimuli, and display increased fungicidal activity (157). As a consequence of this neutrophil hyperactivity, miR-223 mutant mice spontaneously develop inflammatory lung pathology and exhibit exaggerated tissue destruction after endotoxin challenge (157). The latter results indicate that miR-223 is an intrinsic modulator of neutrophil sensitivity, similar to the role proposed for miR-181, which acts as a 'rheostat' controlling T-cell activation (158). Surprisingly, brief exercise alters the miRNA profile in circulating neutrophils in humans (46).
miRNAs IN NK CELLS
NK cells were named after their ability to mediate spontaneous cytotoxicity during the innate immune response. However, it has become clear that they play an equally important role in restricting infection and assisting in the development of adaptive immune responses via their ability to produce cytokines (159,160).
NK cells are a key components of the innate immune system, providing early cellular defense against virus and intracellular pathogens, and contribute to the early detection and destruction of transformed cells (64). Very recently, using mice with conditional deletion of Dicer and DiGerorge syndrome critical region 8 (Dger8), Bezman et al. developed a system for deletion of either Dicer or Dger8 in peripheral NK cells (161). They found that Dicer- and Dger8-deficient NK cells were significantly impaired in survival and turnover, and had impaired function of the immunoreceptor tyrosine-based activation motif (ITAM)-containing activating NK cell receptors. They further demonstrated that both Dicer and Dger8-dependent pathways were indispensable for the expansion of Ly49H+NK cells during mouse cytomegalovirus infection. Thus, their data indicate a critical role for miRNAs in controlling NK cell homeostasis and effector function, with implication for miRNA regulation of diverse aspects of NK cell biology (161). One notable example of an immune activating receptor critically involved in tumor and virus elimination is NKG2D (160). This dominant killer receptor is expressed by NK cells and T-cells, and recognizes stress molecules that are up-regulated on transformed, virus-infected, and other diseased or stressed cells. MICA and MICB are among the stress-induced NKG2D ligands (160). Several stress pathways lead to the up-regulation of NKG2D. However, very little is known concerning the regulation of NKG2D ligand expression in normal conditions. Stern-Ginossar et al. reported a group of endogenous cellular miRNAs that control MICA and MICB expression by binding to MICA and MICB 3'UTR sites (160). Notably, many of the identified cellular miRNAs are over-expressed in various tumors. The authors demonstrated that these miRNAs enable tumors to avoid immune recognition, prompting the proposal of a model in which cellular miRNAs prevent self recognition and promote 'tumor escape' (160).
miRNAs IN NK T-CELLS
NK T-cell (NKT) biology has emerged as a new field of research at the frontier between innate and adaptive immunity (139). Conditional deletion of Dicer has provided insight into the role of miRNAs in controlling the development of B-cells, T-cells, and DCs (84-87,162-165). In one study, dicer deletion early in T-cell development (DN, double negative stage) caused a 10-fold decrease in double positive (DP) and single-positive (SP) thymocyte numbers, although the CD4/CD8 lineage choice was unaffected (163). Dicer deletion at a later stage of T-cell development (DP stage) did not alter the number and composition of mainstream thymocytes, although it resulted in a substantial reduction in thymic CD4+CD25+ Foxp3+nTreg cells (84,87). In the peripheral compartment, the mice showed a modest reduction in CD4+ and CD8+ T cells, Th1 polarization, and increased apoptosis of CD4+ T cells (164). Foxp3-driven deletion of Dicer selectively in Treg cells impairs peripheral homeostasis (85) and suppressor functions (85,86). Recently, Fedeli et al. investigated the invariant NKT (iNKT) cell development in a set of mice in which Dicer was deleted from cortical thymocytes (165). Interestingly, they found that Dicer deletion resulted in a substantial reduction of iNKT cells in the thymus and their disappearance from the periphery, unlike mainstream T-cells. They also found that, without Dicer, iNKT cells were unable to complete their innate effector differentiation and displayed a defective homeostasis due to increased cell death (165). In this study, differentiation and homeostasis of iNKT cells required Dicer in a cell-autonomous fashion. Furthermore, they identified a miRNA profile specific for iNKT cells (165). These data define a critical role of the Dicer-dependent miRNA pathway in the physiology of iNKT cells.
miRNAs IN DCs
DCs are immune cells that effectively link the innate and adaptive arms of the immune system. They are considered a professional antigen-presenting cell population because of their unique capacity to induce the activation and differentiation of naive T-cells at the crossroads of innate and adaptive immunity (69,166). Included in this function is the ability to initiate immunogenic as well as tolerogenic responses from T-cells, a fundamental step in regulating homeostasis. Numerous subpopulations of DCs have been identified to date, including interstitial DCs, dermal DCs, Langerhans cells, plasmacytoid DCs, and inflammatory DCs (166). Importantly, there is accumulating evidence that interactions between DC and Treg cells play a crucial role in the balance between immune response and tolerance (111). Moreover, several lines of experimental evidence suggest that different subset of the functional status of DC are also involved in the prognosis of Treg cell differentiation (111,166). Although relatively little is known about miRNA in DCs, Ceppi et al. recently reported that, in response to LPS, several miRNAs are regulated in human monocyte-derived DCs (167). Among these miRNAs, miR-155 is highly up-regulated during maturation. They also identified the TLR/IL-1 inflammatory pathway as a general target of miR-155. Furthermore, they demonstrated that miR-155 directly controls the level of TAB2, an important signal transduction molecule (167). Their observations suggest that in human mature DCs, miR-155 is part of a negative feedback loop that down-modulates inflammatory cytokine production in response to microbial stimuli (167). More recently, Holmstrom et al. investigated the differential expression of miRNAs in mature DCs (168). Using microarray-based screening, they found that 12 miRNAs were differentially expressed in two DC phenotypes. Of these, four miRNAs (hsa-miR-155, hsa-miR-146a, hsa-miR-125a-6p, and hsa-miR-29a) were validated by real-time PCR and Northern blotting (168).
DCs play important roles in linking the innate and adaptive immune responses. However, few miRNAs regulate the innate response and antigen presenting capacity (APC) function of DCs (69). Calcium/calmodulin-dependent protein kinase II (CaMKII), a major downstream effector of calcium (Ca2+), is an important regulator of the maturation and function of DCs. Liu et al. examined whether miRNA can regulate the innate response and APC function of DCs by targeting CaMKIIα (69). They found that three members of the miRNA-148 family (miR-148a, miR-148b, and miR-152) are negative regulators of the innate response and APC of DCs. They also found that miR-148/152 expression was up-regulated, whereas CAMKII-alpha expression was down-regulated in DCs on maturation and activation induced by TLR agonists. Additionally, they demonstrated that miR148/152 in turn inhibited the production of cytokines including IL-6, IL-12, TNF-α, and IFN-β up-regulation of major histocompatibility (MHC) class II expression and DC-initiated antigen-specific T-cell proliferation by targeting CAMKIIalpha (69). These findings suggest that miRNA-148/152 may act as a fine-tuner in regulating the innate immune response and APC of DCs.
miRNAs IN THE ADAPTIVE IMMUNE RESPONSE
miRNAs are emerging as key regulators of the development and function of adaptive immunity. The contribution of specific miRNAs to the adaptive immune response becomes even more apparent during the effector phases: class -switching and germinal center formation in B-cells, differentiation into functional lineages in T-cells, and activation of antigen-presenting cells through pattern-recognition pathways (169). With the capacity of miRNA to alter the survival and death of T- and B-cells, control over miRNA expression is essential to prevent adaptive immune cells from up-regulated proliferation. miRNA can act both as 'oncomirs' and tumor suppressors, thus dysregulaltion of miRNA in lymphocytes can cause malignancies (66,169). Both developmental and functional roles of miRNA are rapidly being discovered. These studies are either focused on the network-wide role of miRNA or the functions of specific miRNA. The advantage of network-wide studies is the ability to knockout the entire miRNA regulatory network, typically by Cre-Lox mediated excision of genes involved in biogenesis, such as Dicer, Drosha, or Argonaute (84-87). This allows a cell type to be rapidly screened for miRNA-dependent processes in an unbiased fashion, as every miRNA-dependent process should be disrupted. The alternative strategy is to focus on the function of single miRNA, by knockout or overexpression, and determine the affected phenotypes. This strategy is successful at detecting more subtle miRNA functions, in conjunction with mechanistic information (143,151,161,169).
miRNAs IN B-CELLS
B-cell development and differentiation in mice and humans are complex processes that begin in fetal liver and bone marrow with the proliferative expansion of progenitor cells that undergo sequential rearrangements of their antigen receptor repertoire (38). The role of miRNA in B-cell development and B-cell lymphomagenesis is largely unknown (170). One of the key roles of miRNA in B-cells is to allow efficient differentiation in the bone marrow. Removal of the miRNA network through deficiency in Dicer or Ago2 results in a substantial defect in B-cell differentiation from the pre-B-cell stage (162). Chen et al. identified three miRNAs that are specifically expressed in hematopoietic cells and showed that their expression is dynamically regulated during early hematopoiesis and lineage commitment (38). One of these miRNA, miR-181, is preferentially expressed in the B-lymphoid cells of mouse bone marrow. Its ectopic expression in hematopoietic stem/progenitor cells leads to an increase fraction of B-lineage cells in both tissue-culture differentiation assay and adult mice (38). Eis et al. showed that clinical isolates of several types of B-cell lymphomas, including diffuse large B-cell lymphoma (DLBCL), have 10-to 30-fold higher copy numbers of miR-155 than do normal circulating B cells. Similarly, the quantities of bic (B-cell integration cluster) RNA are elevated in lymphoma cells (171). Significantly higher levels of miR-155 are present in DLBCLs with an activated B-cell phenotype. Because patients with activated B-cell-type DLBCL have a poorer clinical prognosis, quantification of this miRNA may be diagnostically useful (171). Evidence suggesting that bic is a primary miRNA transcript containing the mature miR-155 as part of a RNA hairpin is accumulating. Kluiver et al. demonstrated that expression of primary miRNA bic and its derivative miR-155 in Hodgkin's lymphomas, primary mediasinal B-cell lymphoma and DLBCL (172). However, expression of bic and miR-155 is not a common finding in Burkitt lymphoma (173). Mice that are bic-deficient are immunodeficient and fail to generate high levels of class-switched antibody upon immunization with thymus-dependent and thymus-independent antigens (174). In addition, bic-deficient T-cells show skewed differentiation into the Th2 lineage under a variety of in vitro culture conditions. Microarray analysis of bic-deficient B- and T-cells under different conditions has revealed a wide spectrum of targets regulated by an miR-155 and has suggested mechanisms for the regulation of lymphocyte differentiation by a single miRNA (174). Expression of bic and miR-155 in three latency type III EBV (Epstein-Barr virus)-positive Burkitt lymphoma cell lines and in all primary posttranplantation lymphoproliferative disorder cases suggests a possible role for EBV latency type III specific proteins in the induction of bic expression (175).
As mentioned, recent studies have suggested the involvement of miRNA in the initiation and progression of cancer and miR-155, and bic, its host gene, has been reported to accumulate in human B-cell lymphomas (171-173). These reports provide indirect evidence that miR-155 may play a role in B-cell development and lymphomagenesis. The most direct evidence so far for the ability of miRNAs influencing immune responses has come from animal models with both ectopic expression and knockout of candidate miRNA. Costinean et al. reported that the transgenic mice carrying a miR-155 transgene whose expression is targeted to B-cells (Eu-mmu-miR-155) exhibit initially preleukemic pre-B-cell proliferation, evident in spleen and bone marrow, and later develop frank B-cell malignancy (175). miR-155 is expressed by cells of the immune system after activation and has been shown to be required for antibody production after vaccination with Samonella (150). Vigorito et al. showed that B-cells lacking miR-155 generate reduced extrafollicular and germinal center responses and fail to produce high-affinity IgG1 antibodies, and that the transcription factor Pu.1 is a direct target of miR-155-mediated inhibition (150). Rodriguez et al. demonstrated a requirement of bic/miRNA-155 for the function of B-cells (59). Using genetic deletion and transgenic approaches, Thai et al. showed that the evolutionarily conserved miRNA-155 has an important role in the mammalian immune system, specifically in regulating T helper cell differentiation and the germinal center reaction to produce an optimal T-cell-dependent antibody response. miR-155 exerts this control, at least in part, by regulating cytokine production (176). These results also suggest that individual miRNAs can exert critical control over mammalian differentiation processes in vivo (176).
It has become clear over the past few decades that the various phases of B-cell development and response are under distinct programs of transcriptional control, which are activated in the cells by environmental cues. miR-150 controls transcription factor c-Myb expression in vivo and dramatically affects B-cell development (177).
miR-150 is mainly expressed in the lymph nodes and spleen, and is highly up-regulated during the development of mature T- and B-cells (178). In this study, expression of miR-150 was sharply up-regulated at the immature B-cell stage. Over-expression of miR-150 in hematopoietic stem cells had little effect on the formation of T-cells, but the formation of mature B-cells was greatly impaired. Furthermore, premature expression of miR-150 blocked the transition from the pro-B to the pre-B stage (178).
To explore the role of Dicer-dependent control mechanisms in B lymphocyte development, Koralov et al. ablated this enzyme in early B cell progenitor (162). This resulted in a developmental block at the pro- to pre-B-cell transition. Gene-expression profiling revealed a miR17-92 signature in the 3'UTRs of genes upregulated in Dicer-deficient pro-B-cells.
To examine the physiologic effects of an individual miRNA-mRNA interaction in vivo, Dorsett et al. generated Aicda155 mice that harbored a mutation in the putative miRNA-155 binding site in the 3'UTR of activation-induced cytidine deaminase (AID). AID is required for immunoglobulin gene diversification in B lymphocytes (179). They also found that miR-155 can act as a tumor suppressor by reducing potentially oncogenic translocations generated by AID.
To understand the role of miRNAs in B cell function and lymphomagenesis, very recently, Basso et al. generated short-RNA libraries from normal human B cells at different stages of development (naive, germinal center, and memory) and from a Burkitt lymphoma cell line (170). Using a combination of cloning and computational analysis, they identified 178 miRNAs (miRNome) expressed in normal and/or transformed B-cell libraries. Most notably, the B cell miRNome included new 75 miRNAs. Numerous miRNA were expressed in a stage- or transformation-specific fashion in B-cells, suggesting specific functional or pathologic roles (170). These results may provide a resource for studying the role of miRNAs in B-cell development, immune function, and lymphomagenesis. Through concomitant miRNA and mRNA profiling, Zhang et al. demonstrated a potential regulatory role for miRNAs at every stage of the mature B-cell differentiation process (180). In addition, they experimentally identified a direct role for the miRNA regulation of key transcription factors in B-cell differentiation: LMO2 and PRDM 1(Blimp 1). They also profiled the miRNA of B-cell tumors and found that, in contrast to many other malignancies, common B-cell malignancies do not down-regulate miRNA expression (180). Expression of lineage-specific miRNA could correctly predict the lineage of B-cell malignancies in more than 95% of the cases. Thus, their data demonstrate that miRNAs may be important in maintaining the mature B-cell phenotype in normal and malignant B cells (180).
Belver et al. have examined role of miroRNAs in terminal B-cell differentiation by analyzing Cd19-Creki/+Dicer1fl/fl mice (92). In the absence of Dicer, the transitional and marginal zone B-cell compartments were overrepresented and follicular B-cell generation was impaired. Interestingly, microRNA analysis revealed that miR-185, a microRNA overexpressed in follicular cells, dampened B-cell receptor (BCR) signaling through Bruton tyrosine kinase down-regulation. They also found that Dicer-deficient B-cells had a skewed BCR repertoire with hallmarks of autoreactivity, which correlated with high titers of autoreactive antibodies in serum and autoimmune features in females (92). Interestingly, miRNAs prevented the generation of autoimmunity and, importantly, the autoimmune diseases observed in Dicer-deficient animals predominanted in female rather than male mice, very much resembling the profound prevalence of female incidence in various autoimmune disease in humans (13,68). Therefore, the analysis of miRNA expression profiles can prove clinically useful in the diagnosis and prognosis of autoimmune disease.
miRNAs IN CONVENTIONAL T-CELLS
Two studies showed an important role for the miRNA pathway in the development of mature T cells (163,164). In one, Cobb et al. demonstrated that deletion of Dicer at an early stage of T-cell development compromised the survival of αβ lineage cells, whereas the number of γδ-expressing thymocytes were not affected (163). Most remarkably, given that one third of mammalian mRNA target, Dicer seems to be dispensable for CD4/CD8 lineage commitment. Thus, although Dicer seems to be critical for the development of the early embryo, it may have limited impact on the implementation of some lineage-specific gene expression programs (163). In the second study, Muljo et al. generated a conditional allele of dicer-1 in the mouse (164). They reported that specific deletion of dicer-1 in the T-cell lineage resulted in impaired T-cell development and aberrant T helper cell differentiation and cytokine production. A severe block in peripheral CD8+ T-cell development was observed upon dicer-1 deletion in the thymus. However, Dicer-deficient CD4+ T-cells, although reduced in numbers, were viable. These cells were defective in miRNA processing, and upon stimulation they proliferated poorly and underwent increased apoptosis (164). Dicer deletion early in T-cell development induced by a Cre transgene driven by the lck promotor, lckCre, expressed from the double-negative (DN) 2 stage onward, resulted in a sharp reduction of miRNAs by the double-positive (DP) stage and a 10-fold drop in the number of TCR-αβ thymocytes (18,163). Treg cells have a miRNA profile distinct from conventional CD4 T-cells (87). A partial Treg cell-like miRNA profile is conferred by the enforced expression of Foxp3 and, surprisingly, by the activation of conventional CD4 T-cells (87).
Although hundreds of miRNAs are present in the mammalian genome, genetic studies addressing their physiological roles are at an early stage. miR-155 is encoded within an exon of the non-coding RNA known as bic, and high levels of bic expression are induced upon antigen receptor stimulation of B- and T-cells, as well as TLR stimulation of macrophage and DCs (174). Rodriguez et al. reported that mice deficient for bic/miRNA-155 are immunodeficient and display increased lung airway remodeling (59). The authors demonstrated a requirement of bic/miRNA-155 for the function of B-cells, T-cells, and DCs. Transcription analysis of bic/miRNA-155-deficient CD4+ cells identified a wide spectrum of miRNA-155-regulated genes, including cytokines, chemokines, and transcription factors. The study results suggest that bic/microRNA-155 play a key role in the homeostasis and function of the immune system (59). Although no gross defect in myeloid or lymphoid development in bic-deficient mice was observed, protective immunity did appear to be impaired. After intravenous immunization with live attenuated form of the enteric pathogen Salmonella typhimurium (aroA mutant strain), mice were assessed for their ability to resist oral challenge with virulent S. typhimurium (59). Unvaccinated bicm2/m2mice were less readily protected by aroA vaccination, and the majority of mice succumbed to challenge with the virulent strain by 33 days after infection. Thus, immunized bic-deficient mice, unlike wild-type mice, could not be protected by immunization to this pathogen (59). Turner and Vigorito showed that bic-deficient mice were immunodeficient and failed to generate high levels of class-switched antibody upon immunization with thymus-dependent and thymus-independent antigens (174). In addition, bic-deficient T-cells showed skewed differentiation into the Th2 lineage under a variety of in vitro culture conditions. Microarry analysis of bic-deficient B- and T-cells under different conditions has revealed a wide spectrum of targets regulated by an miR-155 and has suggested mechanisms for the regulation of lymphocyte differentiation by a single miRNA (174).
Using genetic deletion and transgenic approaches, Thai et al. showed that the evolutionarily conserved miRNA-155 has an important role in the mammalian immune system, specifically in regulating T helper cell differentiation and the germinal center reaction to produce an optimal T-cell-dependent antibody response (176). miR-155 exerts this control, at least in part, by regulating cytokine production. These results also suggest that individual miRNAs can exert critical control over mammalian differentiation processes in vivo (176). DNA methyltransferase Dnmt1 and DNA methylation are required for the proper expression of certain genes that define fate and determine function in T-cells (181). Banerjee et al. reported that CD4+ T-cells lacking miR-155 exhibit bias towards Th2 differentiation, indicating that the absence of individual miRNA could alter CD4+ T-cell differentiation (182). They also showed that miR-155 induces upon T-cell activation and that it promotes Th1 differentiation when over-expressed in activated CD4+ T-cells by inhibiting IFN-γ signaling.
Neilson et al. observed that individual miRNAs were dynamically regulated during T-cell development, with at least one miRNA or miRNA family overrepresented at each developmental stage, and that miRNA regulation in this developmental pathway is characterized by analog rather than switch-like behavior, with temporal enrichments at distinct stages of development observed against a background of constant and basal expression of the miRNA (183). miR-181a, which is specifically enriched at the CD4+CD8+ (DP) stage of thymocyte development, can repress the expression of Bcl-2, CD69, and the T-cell receptor, all of which are coordinately involved in positive selection (183). T-cell sensitivity to antigen is intrinsically regulated during maturation to ensure proper development of immunity and tolerance. Li et al. observed that increasing miR-181a expression in mature T cells augmented the sensitivity to peptide antigens, while inhibiting miR-181a expression in the immature T-cells reduced sensitivity and impaired both positive and negative selection (158). Moreover, they found that quantitative regulation of T cell sensitivity by miR-181a enabled mature T cells to recognize antagonist - the inhibitory peptide antigens - as agonists. Importantly, higher miR-181a expression correlated with greater T-cell sensitivity in immature T-cells, indicating that miR-181a acts as an intrinsic antigen sensitivity "rheostat" during T-cell development (158).
Interestingly, Johnston et al. found that expression of the transcription factor Bc16 in CD4+ T-cells is both necessary and sufficient for the in vivo differentiation of follicular helper T-cells (Tfh) and T-cell assistance of B-cells in mice. In contrast, the transcription factor Blimp-1, an antagonist of Bc16, inhibits Tfh differentiation and help, thereby preventing B-cell germinal center and antibody responses (184). These findings demonstrate that Tfh cells are required for proper B-cell responses in vivo and that Bc16 and Blimp-1 play central, but opposing, roles in Tfh differentiation.
miRNAs IN TREG CELLS
The discovery of miRNA is one of the major recent scientific breakthroughs and has revolutionized knowledge of gene regulation. Although we are still at a very early stage in understanding their impact of immunity, miRNAs are changing the way we think about the development of the immune system and regulation of immune functions (35,56,185,186). miRNAs are critical for a number of aspects of immune system regulation and function. As mentioned, miRNA are small untranslated RNA species, which have been implicated in the regulation of gene expression essential for organ development, cellular differentiation, homeostasis, and functioning through target mRNA degradation or translational interference (31,35,56,57). To gain functional evidence of the role of miRNA in Treg cell biology, Liston et al. (85) and Zhou et al. (86) recently developed mouse models by depleting Dicer specifically in Foxp3 Treg cell lineage either using mice with Foxp3Cre knock-in allele (85) or Foxp3 GFP-hCre transgenic mice (86). Both mice showed progression of fatal early onset lymphoproliferative autoimmune syndrome during the third week of life, indistinguishable from that observed in Foxp3 mutant mice devoid of Treg cells (84,85,87). Although miRNA-mediated gene expression is critical during B-cell differentiation (162), depletion of miRNA in developing thymocytes does not result in a gross perturbation of T-cell differentiation (163,164). Dicer deletion early in T-cell development induced by a Cre transgene driven by the lck promotor, lckCre, expressed from the double-negative (DN) 2 stage onward, resulted in a sharp reduction of miRNAs by the DP stage and a 10-fold drop in the number of TCR-αβ thymocytes (163,181). However, Cobb et al. (87) and Chong et al. (84) reported that ablation of either Dicer or Drosha, two RNase III enzymes critical for the generation of mature miRNAs, at a stage of the DP thymocyte differentiation, reduces the numbers of Foxp3+ thymocytes and peripheral Treg cells and immune-mediated lesions that develop at 6-mohths-of-age) (84,87). Likewise, Cobb et al. reported a reduction in the efficiency of Foxp3 induction upon stimulation of naive Dicer-deficient T-cells in the presence of TGF-β (87).
Importantly, Dicer-deficient Treg cells lose suppression activity in vivo. As described earlier, in addition to Dicer, Drosha is another RNAseIII enzyme related to miRNA biogenesis (35,84,131,132). By genetic ablation of Drosha within the T cell compartment or specifically within Foxp3+Treg cells, Chong et al. found that, like the Dicer deletion, deletion of Drosha in Treg cells phenocopies mice lacking a functional Foxp3 gene, which further confirmed the critical role of miRNA-dependent regulation in Treg cell development and function, and in preventing spontaneous inflammation and autoimmunity (84). The functional impairment of effector T-cells in CD4Cre-Dicer knockout mice is probably the main reason of the much milder disease found in the CD4Cre-Dicer knockout mouse strain compared to Foxp3Cre-Dicer knockout mice (85,86,163,164). Zheng et al. suggested a potential role of miR-155 in Treg cell formation or function (187). Interestingly, a genome-wide analysis combining chromatin immunoprecipitation with mouse genome tiling array profiling identified Foxp3 binding regions for approximately 700 genes and for an intergenically encoded miRNA (187). The same study reported that a large number of Foxp3-bound genes are up- or down-regulated in Foxp3+ T cells, suggesting that Foxp3 acts as both a transcriptional activator and repressor.
Liston et al. reported that miR-155-deficient Treg cells exhibit impaired homeostasis and proliferative potential, but keep the largely intact in vitro and in vivo suppression functions, suggesting that distinct miRNA species might control different facets of the Dicer-dependent Treg cell phenotype (85). Consistent with the aforementioned observations, using both Tie2Cre- and CD4Cre-mediated Dicer deletion mouse models, Zhou et al. (188) also observed that lack of miRNAs at both stages results in a 2-3-fold decrease in the proportion of Foxp3 Treg cells. These studies have clearly demonstrated a role for miRNA-dependent regulation in the thymic development of CD4+CD25+Foxp3+ Treg cells. However, a general impairment in thymic differentiation observed upon Dicer deletion at the hematopoietic stem cell and early or later T-cell developmental stages mediated by Tie2Cre, IckCre, or CD4Cre obscures the understanding of a specific role for the Dicer-controlled miRNA pathway in Treg cell development and function. Liston et al. (85) found that Dicer deficient Treg cells showed impaired peripheral homeostasis and that Dice-deficient Treg cells from healthy mice were anergic and functional (namely, unable to proliferate in response to TCR stimulation, retaining this distinguishing feature of wild-type Treg cells), although the cells were markedly less efficient on a per cell basis compared with the Dicer-sufficient Treg cells isolated from their wild-type counterparts, similar with that of Treg cells isolated from CD4Cre-Dicer deletion mice (85). However, Dicer-deficient Foxp3+ T cells purified from littermates under conditions of inflammation were completely devoid of suppressor activity and instead showed a robust in vitro proliferative response consistent with the identical autoimmune syndrome in these mice (85). The authors further observed the decreased expression of putative suppressor molecules, including CTLA4, ICOS, IL-10, EBV-induced gene 3 (EBi3), granzyme B, and CD73 in Dicer deficient Foxp3+ Treg cells, in agreement with their marked impairment in suppressor capacity. Interestingly, the Dicer-deficient Foxp3+ Treg cells showed comparable Foxp3 mRNA and protein levels with their wild-type counterpart, supporting the notion that the loss of suppressive capacity was not caused by changes in Foxp3 itself, but rather by low expression of suppressor effecter molecules (85). In contrast, with a Foxp3-GFP-hCre transgenic Treg-specific Dicer deletion mouse model, Zhou et al. (86) showed that mice deleted of Dicer at the time of Foxp3 expression displayed an interrupted Treg cell lineage stability based on their down-regulated Foxp3 expression, which positively correlated with mouse age, even though the mice showed normal thymic Treg cell development at 2-weeks-of-age (86). Moreover, microarray studies also indicated that the majority of Treg cells from Foxp3Cre-Dicer knockout mice expressed altered levels of multiple genes and proteins, largely consistent with a previous study (85), and a significant percentage take on a T helper cell memory phenotype with increased levels of CD127, IL-4, and IFN-γ (86). Cobb et al. also reported that Treg cells have a miRNA profile distinct from conventional CD4 T-cells and that a partial Treg cell-like miRNA profile is conferred by the enforced expression of Foxp3 and, surprisingly, by the activation of conventional CD4 T-cells (87). Depleting miRNAs by eliminating Dicer reduces Treg cell numbers and results in immune pathology (87). Dicer facilitates, in a cell-autonomous fashion, the development of Treg cells in the thymus and the efficient induction of Foxp3 by TGF-β (87). These results suggest that Treg cell development involves Dicer-generated RNAs. Interestingly, the dataset of the study was subjected to significance analysis of microarrays (SAM). Importantly, the authors demonstrated that SAM identified 68 miRNAs that were differentially expressed between nTreg cells and conventional CD4+CD25- T-cells. The authors also showed that 35 miRNAs were preferentially expressed in Treg cells (including miR-223, miR-146, miR-21, miR-22, miR-23a and b, miR-24, miR-214, miR-155, and others) and 33 were down-regulated in Treg cells (including miR-142-5p and -3p, miR-30b, c, e, and members of the Let-7 family). Interestingly, in the same study, the ectopic expression of the Treg cell signature Foxp3 conferred a partial Treg cell miRNA profile (87). Hence, aspects of the Treg cell-specific miRNA profile may be under the direct or indirect control of Foxp3. In addition to the extensive overlap between the miRNA profile of Treg cells and activated T-cells, their analysis has identified miRNAs that are overexpressed by Treg cells but not by activated T-cells, for example miR-223 and miR-146 (87). The thymic differentiation of nTreg cells was compromised in the absence of Dicer and mature miRNAs.
Despite a role of miRNA in the generation of Foxp3+Treg cells both in the thymus and in the periphery, it seems unlikely miRNAs are involved in the regulation of Foxp3 production. Three studies found no evidence in Treg cells in mice with a T-cell specific or Treg-specific Dicer or Drosha deletion (84,85,87). However, Zhou et al. (86) reported that, upon deletion of a conditional Dicer allele mediated by a Foxp3 BAC transgene encoding YFP-Cre, a significant promotion of Dicer-deficient Treg cells became Foxp3 negative, implicating miRNA in Treg lineage stabilization. Furthermore, Liston et al. (85) reported that the miRNA pathway also promoted survival and proliferative potential of Treg cells, similar to its role in T- and B-cell lineages.
In addition to its role in Treg development and homeostasis, miRNA-dependent regulation of gene expression is also critical to controlling Treg cell function. It is interesting to note that depletion of miRNA within the Treg cell lineage can result in a fatal autoimmunity indistinguishable from that in Treg cell-deficient mice (84-86). Interestingly, the suppressor capacity of Dicer-deficient Treg cells was maintained, albeit at a markedly reduced level, under noninflammatory conditions (84-86). However, Liston et al. (85) showed that in inflammatory settings, these cells entirely lost the suppressor capacity despite a marked increase in their numbers and activation. These observations may implicate miRNAs as key guardians of a stable Treg suppressor program under inflammatory conditions.
Treg-specific Dicer and Drosha ablation studies have raised a question as to the identity of specific miRNAs regulating distinct aspects of Treg cell biology. Cobb et al. first demonstrated differential miRNAs expression in Foxp3+ Treg cells in 2006 (87). Furthermore, many Treg-specific miRNAs are expressed in Treg cells in a Foxp3-dependent manner. Among the Foxp3-dependent miRNAs, miR-155 is directly regulated by Foxp3 (98,151,187). Although dispensable for Treg differentiation and suppressor function, Foxp3-driven miR-155 up-regulation is critical for heightened responsiveness of Treg cells to their key survival and growth factor, IL-2.
At a mechanistic level, miR-155-mediated control of Treg cell homeostasis is mediated through the targeting of SOCS1, a negative regulator of IL-2 signaling. Thus, constitutively high expression of miR-155 driven by Foxp3 ensures efficient STAT5 phosphorylation in the presence of limiting amounts of IL-2 and, thus, fitness of Treg cell subset in a competitive environment (151). This finding not only provides the first example of a single miRNA controlling Treg homeostasis but also demonstrates that different miRNAs affect distinct facets of Treg cell biology. Treg cells limit the pathogenic immune response to self-antigens and foreign antigens. An essential role for miRNA in the maintenance and functions of Treg cells, revealed by the Treg cell-specific Dicer ablation, raises a question as to a specific miRNA contribution. Lu et al. (151) found that Foxp3 controlled the elevated miR-155 expression required for maintaining Treg cell proliferative activity and numbers under nonlymphopenic conditions. Moreover, they also found that miR155 deficiency in Treg cells resulted in increased SOCS1 expression accompanied by impaired activation of STAT5 transcription factor in response to limiting amounts of IL-2. Their study suggests that Foxp3-dependent regulation of miR-155 maintains the competitive fitness of Treg cells by targeting SOCS1, and provides experimental support for a proposed role for miRNAs in ensuring the robustness of cellular phenotypes (151). This finding not only provides the first example of a single miRNA controlling Treg homeostasis but also demonstrates that different miRNAs affect distinct facets of Treg cell biology.
Lu et al. (151) provided experimental evidence showing that: 1) Foxp3 regulates miR-155 expression in Treg cells, 2) miR-155 deficiency results in a reduction of Treg cell numbers, 3) diminished proliferative potential of miR155-deficient Treg cells, 4) miR-155 maintains competitive fitness of Teg cells, 5) attenuated IL-2 signaling in miR-155-deficient Treg cells, and 6) miR-155 maintains Treg cell homeostasis by limiting SOCS1 protein expression; SOCS 1 protein amounts are low in miR-155-sufficient Treg cells. In contrast, miR-155-deficient Treg cells exhibit an approximately 5-fold increase in the amount of SOCS1 (151). Recently, Kohlhass et al. (189) investigated the contribution of miR-155 to Treg development and function. They reported that mice deficient in bic/miR155 showed reduced numbers of Treg cells in the thymus and spleen. They also found that Treg cell development required bic/miR155, but that it was dispensable for Treg cell proliferation or survival in the periphery. Despite the lower numbers of Treg cells, their suppressor function in vitro remained intact. Furthermore, no signs of spontaneous inflammatory bowel disease (IBD) were observed in young or aged miR-155-deficient mice, and miR-155-deficient CD4+CD25+ T cells were able to prevent colitis induced by the adoptive transfer of CD4+CD45RBhigh T cells into lymphopenic hosts (189). The results indicate that miR-155 contributes to Treg cell development, but its absence does not overtly compromise Treg function in vitro and in vivo.
Human umbilical cord blood (UCB) contains a distinct population of CD25+Treg cells that are less heterogeneous than in adult peripheral blood. Indeed, except for fetal infection, the majority of human cord blood CD4+CD25+ T cells express Foxp3 (190). Thus, cord blood is a convenient source for studies aimed at understanding human nTreg, because, in contrast to adult blood, as they are less contaminated by CD25+-activated non-Treg T cells. Moreover, while Foxp3 expression is not sufficient to identify Treg cells in adult human blood, it remains a marker of choice in UCB lymphocytes from healthy newborns and in adult peripheral blood Treg cells provided they are distinguished from activated T cells (190). miRNAs signatures have been described in mouse Treg cells (84-87,191). Studies in humans have been done to identify a human miRNA Treg signature in healthy volunteers and patients (186,190,192-195). Very recently, Redouane et al. investigated human nTreg using UCB (190) and identified 10 miRNAs that were differentially expressed between nTreg and naive CD4+CD25+ T cells. Eight miRNAs were preferentially expressed in nTreg (miR-425-Sp, miR-181c, miR-21, miR-374, miR-586, miR-340, miR-26b, and miR-491) and two were selectively down-regulated in nTReg (miR-31 and miR-125a) using the TaqMan Low Density Array (TLDA) technique (190). Differential miRNA expression was then validated by real-time PCR. A nTreg miRNA signature was defined, consisting of five statistically differentially expressed miRNAs (miR21, miR-31, miR-125a, miR-181c, and miR-374). These miRNAs were detected using the TLDA technique and confirmed by quantitative PCR (190). Three of the five miRNAs were over-expressed (miR-21, miR-181c, and miR-374) and the remaining two were under-expressed (miR-31 and miR-125a). They also identified a functional target sequence for miR-31 in the 3'UTR of Foxp3 mRNA (190). Using lentiviral transduction of fresh cord blood T cells, the authors demonstrated that miR-31 negatively regulated Foxp3 expression by binding directly to its potential target site in the 3'UTR of Foxp3 mRNA (190). The authors also demonstrated that miR-21 acted as a positive, though indirect, regulator of Foxp3 expression. Transduction of the remaining three miRNAs had no direct effect on Foxp3 expression or on the phenotype and will remain the subject of future investigation. Taken together, the study not only found and validated a miR signature for human nTreg for the first time, but also unveiled some of the mechanisms by which this signature may be involved in the control of the Foxp3 levels in nTreg. This opens several possibilities for the future, i.e. to qualitatively investigate induced Treg in several circumstances, healthy volunteers, and patients suffering from cancer and immune disorders. These experiments, coupled with quantitative measurements in these different situations, could provide new insights into the defective immune response in malignancies, immune disorders, infections and transplantation.
Hussein et al. reported that histone deacetylase inhibitors increased Foxp3 expression in T cells (192). They therefore decided to investigate in non-Treg CD4-positive cells the mechanisms by which an a specific opening of the chromatin could lead to an increased Foxp3 expression. They focused on binding of potentially activating transcription factors to the promoter region of Foxp3 and on modifications in the five miRNAs constituting the Treg cell signature. Valproate treatment also induced binding of E-twenty six family-1 (Ets-1) and Ets-2 to the Foxp3 promoter and acted positively on its expression, by increasing the acetylation of histone H4 lysines (192). Vaproate also induced the acquisition of the miRNA Treg cell signature (192). To elucidate whether the changes in the miRNA expression could be due to the increased Foxp3 expression, the authors transduced these non-Treg cells with a Foxp3 lentiviral expression vector, and found no changes in miRNA expression. Therefore, the modification in their miRNA expression profile was not due to an increased expression of Foxp3 but resulted directly from histone deacetylase inhibition. Rather, the increased Foxp3 expression results from the additive effects of Ets factor binding to its promoter, and the change in expression level of miR-21 and miR-31. The authors noted that valproate treatment of human non-Treg cells confers a molecular profile similar to that of their regulatory counterparts (192). Their findings strongly suggested that valproate treatment increases Foxp3 mRNA and protein levels in CD4+, CD25- T cells and that valproate-treated UCB CD4+, CD25-T cells transiently adopt a nTreg miRNA expression signature. Taken together, the data indicate that CD4+CD25-UCB T cells transiently adopt a nTreg cell-like miRNA signature upon valproate treatment (high Foxp3 expression, low miR-31 and miR-125a expression, and high level of miR-21, miR-181c, and miR-374). Ets1 and Ets-2 stimulate the Foxp3 promoter.
Xiao et al. reported the requirement of Treg cells for the maintenance of immune homeostasis and that dysfunction of Treg cells leads to fatal autoimmunity in humans and mice (30). Conversely, the activation of different classes of Treg cells operative systemically and within the cancer microenvironment can suppress host anti-tumor immune responses and promote tumor progression. Therefore, the development of new therapeutic approaches to regulate the activity of Treg cells may have considerable clinical potential. It is well-known that Foxp3 is a key transcriptional regulator of Treg development and function (14). The activity of Foxp3 is regulated by acetylation, a process catalyzed distinct types of histone/protein acetyltransferases (HATs) that regulate the functions of many transcription factors, independent of Foxp3, as well as non-histone proteins, in addition to their effects on chromatin accessibility (30). Interactions between Foxp3 and these enzymes determine the suppressive function of Foxp3. Clearly, small molecules targeting these enzymes are candidates for the regulation of Treg function in vaccines and tumor therapies (30).
Multiple sclerosis (MS) is a chronic inflammatory response against constituents of the CNS. Treg cells play a key role in the autoimmune balance and their improper function may facilitate the expansion of autoaggressive T cell clones (13,66). Involvement of miRNAs in autoimmune disorders and their loss-of-function in immune cells facilitates systemic autoimmune disorders (66,99). De Santis et al. (143) found 23 human miRNAs differentially expressed between CD4+ CD25high bona fide Treg cells from MS patients versus healthy donors, but, conversely, among the deregulated miRNAs, members of the miR-106b-25 were found down-regulated in MS patients when compared to healthy donors in CD4+ CD25highCD127dim/- Treg cells. More interestingly, the ratio between Treg/Teff showed an enrichment of these miRNA in Treg cells derived from patients if compared to healthy controls (143). The data suggested that the abnormal expression of miRNA in Treg cells might play a role in the pathogenesis of MS.
Cobb et al. raised a question as to how individual miRNAs present in increased amounts in Treg cells contribute to distinct aspects of their homeostasis and function (87). Recently, it has been reported that miR-155, a well-known onco-miRNA (cancer-associated miRNA), is constitutively expressed in high amounts in Treg cells in a Foxp3-dependent manner, whereas T cells lacking Foxp3, B cells, and myeloid cells are transiently up-regulated miR-155 upon activation (149,155,176). Lu and Rudensky (196) also reported that miR-155 confers heightened responsiveness of Treg cells to their key survival and growth factor, IL-2, thereby maintaining their numbers in a competitive environment. However, miR155 is largely dispensable for Treg suppressor function (196).
Like miR-155, miR-146a is highly expressed in Treg cells. It has been demonstrated that miR-146a, which is up-regulated during viral infection, is a negative regulator of the retinoic acid-inducible gene (RIG)-I-dependent antiviral pathway by targeting TRAF6 (TNFR-associated factor), IRAK1 (IL-1R-associated kinase 1), and IRAK 2 (153,154). Curtale et al. showed that miR-146 is low in human naive T cells and is abundantly expressed in human memory T cells (169). They also showed that miR-146a modulated activation-induced death domain (AICD) by acting as an antiapoptotic factor and that FADD is a target of miR-146. Furthermore, they demonstrated that miR-146a enforced expression impairs both activator protein 1 (AP-1) activity and IL-2 production induced by TCR engagement. More recently, Lu et al. (197) explored a role for miR-146a in Treg cells and reported that miR-146a, which is prevalently expressed in Treg cells, is essential for the ability of Treg cells to restrain IFN-γ-mediated pathogenic Th1 responses and associated inflammation. They also found that in Treg cells, miR-146a-mediated down-regulation of STAT1, a key transcription factor required for Th1 effector cell differentiation, was necessary for Treg cell ability to suppress Th1 response. Likewise, heightened STAT1 activation in Treg cells subjected to a selective ablation SOCS1, a key negative regulator of STAT1 phosphorylation downstream of the IFN-γ receptor, was associated with analogous Th1-mediated pathology (197). Furthermore, they found that relieving negative regulation of STAT1 activation in Treg cells using an alternative genetic approach resulted in a breakdown of immune homeostasis similar to that observed in mice harboring miR-146a-deficient Treg cells. Thus, their results suggest that specific aspects of Treg cell suppressor function are controlled by a single miRNA and that an optimal range of STAT1 activation is important for Treg-mediated control of Th1 responses and associated autoimmunity.
The generation of mice with a conditional deletion of Dicer or Drosha in Treg cells has shown a requirement for the miRNA pathway in Foxp3+Treg cells (84-86,151,189). These mice develop a lethal autoimmune inflammatory disease, consistent with impaired development or function of Treg cells homeostasis and overall survival, and this is thought to involve the direct targeting of SOCS1 (151,189). However, because the absence of miR-155 did not reproduce the severe disease that occurs in mice with a conditional deletion of Dicer, additional miRNAs are probably involved in Treg cell biology. Several other miRNAs are expressed by Treg cells and await functional assessment (87,186,193).
Phosphodiesterases, the enzyme that accelerate the turnover of cAMP, are up-regulated in activated helper T cells and down-regulated in Treg cells (98,187). Interestingly, Huang et al. showed that miR-142-3p regulates the production of cAMP by targeting adenyl cyclase (AC) 9 mRNA in CD4+CD25- T cells and CD4+CD25+ Treg cells (191). Moreover, they showed that miR-142-3p limits the level of cAMP in CD4+ CD25- T cells by inhibiting AC9 production, whereas Foxp3 down-regulates miR-142-3p to keep the AC9/cAMP pathway active in CD4+CD25+ Treg cells. These findings reveal a new molecular mechanism through which CD4+CD25+ Treg cells contain a high level of cAMP for their suppressor function, and also suggest that the miRNA controlling AC expression might restrict the final level of cAMP in various types of cells. Interestingly, Bopp et al. reported that the suppressive activity of nTreg cells is abolished by a cAMP antagonist as well as by a gap junction inhibitor, suggesting an unexpected role for a classical second messenger in combination with an ubiquitous system of intercellular communication in nTreg cell-mediated suppression (198). Of note, the expression of miR-142-3p was recently shown to be repressed by Foxp3, leading to increased production of cyclic AMP and suppressor function of Treg cells (191).
Asirvatham et al. studied 613 genes that regulate immunity and identified 285 genes as miRNA targets (199). Interestingly, they found that major targets include transcription factors, cofactors and chromatin modifiers upstream factors, such as ligand and receptors (cytokines, chemokines and TLRs) were, in general, non-targets. About 10% of immune genes were 'hubs' with eight or more different miRNAs predicted to target 3'UTRs. Hubs were focused on certain key immune genes, such as BCL6, SMAD7, BLIMPT, NFAT5, EP300, and others (199). Moreover, multiple components involved in the generation and effector functions of miRNAs (Dicer and Argonautes) were themselves miRNA targets suggesting that a subset of miRNA may indirectly control their own production as well as other miRNAs (199).
Treg cells are the main mediators of dominant tolerance. Their mechanisms of action and applications are subjects of considerable debate currently. Rouas et al. (193) investigated human nTReg and identified a signature composed of five miRNA (miR-21, miR-31, miR-125a, miR-181c, and miR-374). Among those, two were considerably under-expressed (miR-31 and miR-125a). The authors also identified a functional target sequence for miR-31 in the 3'UTR of Foxp3 mRNA. Using lentiviral transduction of fresh cord blood T cells, the authors demonstrated that miR-31 and miR-21 affected Fosp3 expression by binding directly to its potential target site in the 3'UTR of Foxp3 mRNA. Additionally, they demonstrated that miR-21 acted as a positive, although indirect, regulator of Foxp3 expression.
Recent studies suggest a role for Dicer and Drsha-controlled miRNA pathways in Treg cell-mediated immune tolerance (84-87) and of miRNAs in the development and function of Treg cells (86,189). Treg cells from normal animals display a specific miRNA profile (84-87). However, it is unclear whether Dicer and miRNAs play a role in eliciting Treg cell defect in humans and mice with autoimmune disease such as systemic lupus erythematosus (SLE). SLE is a chronic autoimmune disease characterized by loss of tolerance to self-antigens and activation of autoreactive T cells. Treg cells play a critical role in controlling the activation of autoreactive T cells. More currently, Divekar et al. (186) investigated mechanisms of potential Treg cell defect in SLE using MRL-Faslpr/lpr (hereafter referred to as MRL/lpr) and congenic Fas-intact MRL-Fas+/+ (MRL+/+) mouse models. Paradoxically, they found that a significant increase in CD4+CD25+Foxp3+ Treg cells, albeit with an altered phenotype and with a reduces suppressive capacity, in the lymphoid organs of MRL strains, and a profound reduction in Dicer expression and an altered miRNAs profile in MRL/lpr Treg cells. Despite having a reduced level of Dicer, MRL/lpr Treg cells exhibited a significant overexpression of several miRNAs, including let-7a, let 7f, miR-16, miR-23a, miR-23b, miR27a, and miR-155. Surprisingly, autoimmune disease development in MRL/lpr mice was associated with significantly increased miR-155, despite a profound reduction in Dicer expression (186). They also demonstrated the first example of an acquired deficiency of Dicer, which occurs spontaneously in MRL/lpr mice just prior to the onset of inflammatory disease. Thus, genetic deficiency of Dicer in knockout mice as well as its acquired deficiency in MRL/lpr mice is associated with Treg abnormalites and development of autoimmune disease, suggesting an important role of Dicer in Treg cell function and prevention of autoimmunity (186). Indeed, the Dicer-controlled miRNA pathway is important in the control of autoimmunity (84-87). Using computational and experimental approaches, Divekar et al. further identified miR-155 as regulating an altered phenotype of Treg cells in SLE (186). Together, these data suggest a role of Dicer and miR-155 in conferring Treg cell defect in SLE. Furthermore, simultaneous appearance of Dicer insufficiency and miR-155 over-expression in disease mice suggests a Dicer-independent alternative mechanisms of miRNA under inflammatory conditions (186). In Treg cells of diabetic patients, interestingly, Hezova et al. found significantly increased expression of miRNA-510 and decreased expression of both miRNA-342 and miRNA-191 (194). When comparing Treg cells and T cells, it was revealed that Treg cells had significant higher expression of miR-146a and lower expression of eight specific miRNAs (20b, 31, 99a, 100, 125b, 151, 335, and 365). This may be the first study demonstrating changes in miRNA expression profiles occurring in Treg cells of type 1 diabetic patients and a miRNAs of adult Treg cells.
Freier et al. analyzed the behavior of Treg cells in the context of a stress-induced activation of the adaptive immune response in healthy young males (195). The authors found that acute psychological stress caused a concomitant decrease in CD4+FOXP3 Treg cells and in CD4+ T cells expressing Treg cell-related effector molecules CTLA-4 and latency associated peptide (LAP). In addition, they observed β1-adrenergic and glucocorticoid α receptor to be over-expressed in Treg cells, suggesting that these molecules might mediate stress-related effects on Treg cells (195). It has been reported that inhibiting components of the adaptive immune response, like Treg cells, are down-regulated during a stress-induced activation of the adaptive immune response. Interestingly, however, in chronic stress, this scenario might result in an exacerbation of inflammatory conditions, such as autoimmune diseases (195).
The potential to therapeutically regulate miRNA levels may offer new avenues for cancer treatment and possibly in regulating the immune system. Inhibiting oncogenic miRNAs or reintroduction of tumor suppressor miRNAs may serve as useful strategies to treat cancer. A recent study clearly demonstrated that reintroduction of tumor suppressor miRNAs can cause apoptosis or senescence in malignant cells and provide new avenues for development this potential in mouse models of hepatocellular carcinoma (200). Clinical trials evaluating therapy based on miRNA inhibition or over-expression (42). Within the next several years, we will know if miRNA-based therapeutics, alone or in combination with other modalities, will be clinically useful treatments for various cancers and immune system disorders (42).
POSSIBLE CLINICAL APPLICATION OF miRNAs
As mentioned, miRNAs control various activities of the immune system and different stage of hemaotopoietic development, and their mis-expression is the cause of various blood malignancies. Certain miRNAs have oncogenic activities, whereas others have the potential to act as tumor suppressors (35,36,57,60,77), suggesting their contribution to cancer development and progression. Because miRNAs control fundamental process such as differentiation, cell growth, and death, the study of the role of miRNAs in human neoplasms holds great promise for novel forms of therapy (138). Therefore, miRNA profiling is used to create signature for a variety of cancers, indicating that the profile will help further establish molecular diagnosis, prognosis, and therapy using miRNA (60,77,86,128). miRNA expression profiles can be used to distinguish normal B-cells in patients with chronic lymphocytic leukemia (CLL). A unique miRNA signature is associated with prognostic factors and disease progression in CLL (201). Large high-throughput studies in patients revealed that miRNA profiling has the potential to classify tumors and predict patient outcome (74). miRNA deficiencies or excess have been correlated with a number of clinically important disease ranging from myocardial infarction to cancers (99). Yu et al. investigated whether miRNA expression profiles can predict the clinical outcome of NSCLC (non-small-cell lung cancer) patients (93). They identified a five-miRNA signature for the prediction of treatment outcome of NSCLC: two miRNAs (hsa-miR-221 and hsa-let-7a) were protective and the other three miRNAs (hsa-miR-137, hsa-miR-372, and hsa-miR-182*) were risky. They also reported that patients with high-risk scores in their miRNA signatures had poor overall and disease-free survivals compared to the low-risk-score patients, and that this miRNA signature was an independent predictor of the cancer relapse and survival of NSCLC patients (93). Multivariate regression analysis showed that the miRNA signature is independent from stage or histology and that the miRNA signature can predict patient survival within cancer stages and histological subgroups of NSCLC patients (93). This may have prognostic or therapeutic implications for the management of NSCLC patients.
The potential value of miRNAs as prognostic and predictive biomarkers in cancer is elegantly highlighted in the recent work. Schetter et al. (202) compared miRNA expression patterns in colon adenocarcinoma and adjacent normal tissue using a test set and validation cohorts. They found that specific miRNA signature distinguished colon cancer from normal colon, and a subset of miRNAs was further demonstrated to exhibit prognostic marker of poor survival (202). In addition, they reported that high miR-21 expression predicted worse survival in treated colon cancer patients and poor responsiveness to adjuvant chemotherapy (202). Beyond prognosis and prediction, miRNAs associated with neoplastic transformation may mediate pathophysiological mechanisms underlying tumorigenesis (91,203). Waldman and Terzic also reported that the role of miRNAs in tumorigenesis underscores their value as a mechanism-based therapeutic target in cancer (90). Similarly, unique patterns of altered levels of miRNA production provide fingerprints that may serve as molecular biomarkers for tumor diagnosis, classification, prognosis of disease-specific outcomes, and prediction of therapeutic responses (90). Many miRNAs with oncogenic activity in hematologic malignancies have been reported. These so-called oncomirs such as the miR-17-92 cluster, miR-21, and miR-155 are often over-expressed in malignant tissues (204). For example, the miR-17-92 cluster is a target of genomic amplification 13q31 that occurs in Burkitt's lymphoma, diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma, and follicular lymphoma (2004). In addition to the miR17-92 cluster, miR-155 is one of the oncomirs that has been well characterized in the adaptive immune system in both healthy development and malignancy, including B-cell lymphoma, Hodgkin's lymphoma, DLBCL, and Burkitt's lymphoma. Expression of primary miRNA BIC and its derivative miR-155 have been demonstrated in Hodgkin's lymphoma, primary mediastinal B-cell lymphoma (PMBL), and DLBCL (205).
In contrast to miRNA oncogene function described above, other miRNAs function as potent tumor suppressors. The loss of such miRNAs is frequently associated with T-cell and B-cell lymphoma and leukaemia (206). One example is miR-15a and miR-16-1, two miRNAs that are clustered in the 13q14 region. In more than 50% of CLL patients, this region is deleted (206). Similarly, miR-29 and miR-181 are down-regulated in CLL, and have both function as tumor suppressors (207). These miRNAs normally suppress proliferation by targeting pro-survival and pro-proliferation genes including BCL-2, TCL1, MCL1, and CDK6 (206,207). Interestingly, a search for general regulators of cancer metastasis has yielded a set of miRNAs for which expression is specifically lost as human breast cancer cells develop metastatic potential (82). Interestingly, Tavazoie et al. showed that restoring the expression of these miRNAs in malignant cells suppresses lung and bone metastasis by human cancer cells in vivo (199). Of these miRNAs, miR-126 restoration reduces overall tumor growth and proliferation, whereas miR-335 inhibits metastatic cell invasion. Expression of miR-126 and miR-335 is lost in the majority of primary breast tumors from patients who relapse, and the loss of expression of either miRNA is associated with poor distal metastasis-free survival. Thus, miR-335 and miR-126 are metastasis suppressor miRNAs in human breast cancer (208).
As mentioned, miRNAs have been implicated in several carcinogenic process, where they can act either as oncogenes or as tumor suppressors (90,204,206). This is the case in lung cancer (the leading cause of cancer death in Western countries), in which about 40-45 miRNAs have been found to be aberrantly expressed, thereby constituting a specific miRNA signature (76). The different expression profiles of miRNAs in lung cancer, and the stability of miRNA in serum (95), all together implicate them as new potentially clinical biomarkers for diagnosis and prognosis (49). Moreover, miRNAs may serve as either novel potential targets acting directly as oncogene (e.g., miR-17-19 cluster), or directly as therapeutic molecules working as tumor suppressor genes, such as the let-7 family (49). Some of these miRNAs can play an important role in lung carcinogenesis. Changes in the expression profiles of miRNAs have been observed in colorectal cancer (CRC) and several investigators have also described the ability of miRNAs expression profiles to predict prognosis, diagnosis, and response to selected treatments in CRC patients (103). The occurrence of miRNAs has been repeatedly observed in serum and plasma, and miRNA as novel minimally invasive biomarkers indicate the reasonable sensitivity for CRC detection (103).
Therapeutic strategies based on modulation of miRNA activity hold great promise due to the ability of these small RNAs to potently influence cellular behavior. Kota et al. investigated the efficacy of a miRNA replacement therapy for liver cancer (200). They found that hepatocellular carcinoma (HCC) cells exhibit reduced expression of miR-26a, a miRNA that is normally expressed at high levels in diverse tissues. Expression of this miRNA in liver cancer cells in vitro induces a G1 cell-cycle arrest associated with direct targeting of cyclines D2 and E2 (200). Importantly, they also showed that systemic administration of this miRNA in a mouse model of HCC using adeno-associated virus (AAV) results in inhibition of cancer cell proliferation, induction of tumor-specific apoptosis, and dramatic protection from disease progression without toxicity, indicating that AAV-mediated miR-26a delivery is very powerfully effective. These findings provide proof-of-concept support for systemic delivery of tumor-suppressing miRNAs as a powerful and highly specific anti-cancer therapeutic modality (200).
More recently, it has been reported that serum and other body fluids contain sufficiently stable miRNA signatures (62,95,144,146,209). Thus, the profiles of circulating miRNAs have been explored in a variety of studies aiming at the identification of novel non-invasive biomarkers, particularly functions of circulating miRNA in the serum miRNAs are currently being extensively explored for their potential as non-invasive diagnostic tumor markers (95,144,146,209). Tumor specific circulating miRNAs may improve cancer diagnosis and prognosis, since several promising miRNAs have already been described as non-invasive biomarkers for different tumor entities (95).
As for miRNAs-regulating a change of heart, similar delivery strategies may enable specific administration of miRNA to the heart. miRNAs regulate cardiovascular structure and function (101). Overexpression of miR-133 blocked cardiac hypertrophy in Akt transgenic mice predisposed to left ventricular hypertrophy (LVH), whereas wild-type mice developed LVH in response to administration of a miR-133 antisense oligonucleotide (ASO) inhibitor. However, LVH was not observed in miR-133-deficient mice (101). Clearly, miRNAs play a significant role in cardiovascular development and disease. Additionally, miRNAs are important for regulating cardiomyocyte self-renewal and differentiation, as well as for normal cardiac structural integrity. Results like these in disease models suggest that inhibiting inappropriately expressed miRNAs with the use of ASOs and, perhaps replacing missing miRNAs, can be used to treat human cardiac disease (101). Identification of miRNAs and target genes that contribute to adult cardiac pathology is likely to suggest new targets for therapy. Obstacles encountered previously with gene therapy and antisense drugs remain an impediment to delivering miRNAs, but preclinical studies for antisense miRNA inhibitors have been promising. Similarly, both preclinical studies and phase II human trials have been encouraging, suggesting that miRNA replacement strategies and miRNA inhibitors will find their way into clinical use in the not too distant future (94,101).
Drug-induced liver injury is a frequent side effects of many drugs, constituting a significant threat to patient health, and has an enormous economic impact on health care expenditures. The most commonly use diagnostic test for liver injury is to determine the activity of certain hepatocelluar enzymes, aspartate aminotransferase (AST or SGOT) and alanine aminotranserase (ALT or SGPT), in the blood. However, this information is unreliable (210). Numerous efforts have been made to identify reliable and predictive markers to detect the early signs of drug-induced injury to the liver, one of the most vulnerable organs in the body. Wang et al. demonstrated that specific miRNA species, such as miR-122 and miR-192, are both enriched in the liver tissue and plasma in the mouse as model system., indicating the potential of using specific circulating miRNA as sensitive and informative biomarkers for drug-induced liver injury (106). Recently, discovery of miRNA in the body fluids (such as blood, plasma, serum, and saliva), has suggested the potential for miRNA-based biomarkers in the detection of cardiovascular diseases such as myocardial damage, coronary artery disease (144,145,209), cancer (146,148,211-214), and oral disease (147), indicating that miRNA may be an excellent blood-based biomarker. These results, although preliminary, strongly suggest that a more comprehensive study of circulating miRNAs and their association with various physiopathological conditions may lead to another dimension in the discovery of biomarkers in the blood for many physiological and pathological conditions. It has been reported that lower complexity, no post-processing modification, synthetic high-affinity "capsule" reagent, tissue-restricted expression profile, and "amplicable" signals make circulating miRNA ideal candidates as biomarkers to reflect various physiopathological conditions in the body (210).
CONCLUDING REMARKS
It is becoming clear that miRNA are being identified as key regulators of immune cell development and function, as well as disease pathogenesis. miRNA signatures have become one of the most fascinating interests in current biology and medical science. Emerging evidence suggests that miRNAs play a key role in the regulation of immunological functions including innate and adaptive immune responses, development and differentiation of immune cells, and the prevention of autoimmunity. It is also now evident that aberrant miRNA expression in the immune system is sufficient to cause disease, and so proper regulation of miRNA expression seems to be crucial for disease prevention.
To gain functional evidence of the role of miRNA in Treg cell biology, several investigators recently have made mouse models depleting Dicer specifically in Foxp3 Treg cell lineage, either using mice with Foxp3Cre knock-in allele (85) or using Foxp3 GFP-hCre transgenic mice (86). Both mice showed progression of fatal early onset lymphoproliferative autoimmune syndrome indistinguishable from that observed in Foxp3 mutant mice devoid of Treg cells. Ablation of either Dicer or Drosha results in reduced numbers of Foxp3+ thymocytes and peripheral Treg cells and immune-mediated lesions developing at 6-months-of-age. Dicer deficient Treg cells showed impaired peripheral homeostasis. Decreased expression of putative suppressor molecules, including CTLA4, ICOS, IL-10, EBV-induced gene 3 (EBi3), granzyme B, and CD73 in Dicer deficient Foxp3+ Treg cells has also been described.
Treg cells have a miRNA profile distinct from conventional CD4 T cells and a partial Treg cell-like miRNA profile is conferred by the enforced expression of Foxp3 and, surprisingly, by the activation of conventional CD4 T cells. Depleting miRNAs by eliminating Dicer reduces Treg cell numbers and results in immune pathology. Dicer facilitates the development of Treg cells in the thymus and the efficient induction of Foxp3 by TGF-β. These results suggest that Treg cell development involves Dicer-generated RNAs.
At a mechanistic level, miR-155-mediated control of Treg cell homeostasis is mediated through the targeting of SOCS1. miR-155 deficiency in Treg cells results in increased SOCS1 expression accompanied by impaired activation of STAT5 transcription factor in response to limiting amounts of IL-2.
A large number of studies have reported 1) Foxp3 regulates miR-155 expression in Treg cells, 2) miR-155 deficiency results in a reduction of Treg cell numbers, 3) diminished proliferative potential of miR155-deficient Treg cells, 4) miR-155 maintains competitive fitness of Teg cells, 5) attenuated IL-2 signaling in miR-155-deficient Treg cells, 6) miR-155 maintains Treg cell homeostasis by limiting SOCS1 protein expression; SOCS 1 protein amounts are low in miR-155-sufficient Treg cells, whereas miR-155-deficient Treg cells exhibit an approximately 5-fold increase in the amount of SOCS1 (151).
More recently, a human miRNA Treg signature has been explored in healthy volunteers and patients. A variety of human miRNA Treg signatures have been reported (186,190,192-195). Eight miRNAs are preferentially expressed in nTreg (miR-425-Sp, miR-181c, miR-21, miR-374, miR-586, miR-340, miR-26b, and miR-491) and two are selectively down-regulated in nTReg (miR-31 and miR-125a). This opens several possibilities for the future, i.e. to qualitatively investigate induced Treg in several circumstances, healthy volunteers, and patients suffering from cancer and immune disorders.
Like miR-155, miR-146a is highly expressed in Treg cells. miR-146a also modulates activation-induced death domain (AICD), acting as an antiapoptotic factor and that FADD (Fas-associated protein with death domain) is a target of miR-146.
Experimental findings have revealed new molecular mechanisms through which CD4+CD25+ Treg cells contain a high level of cyclic AMP (cAMP) for their suppressor function. Of note, the expression of miR-142-3p was recently shown to be repressed by Foxp3, leading to increased production of cyclic AMP and suppressor function of Treg cells.
As for clinical application of miRNA and Treg cells, certain miRNAs have oncogenic activities, whereas others have the potential to act as tumor suppressor, suggesting their contribution to cancer development and progression. Recent studies of the role of miRNAs in human neoplasms hold great promise for novel forms of therapy. Therefore, miRNA profiling is used to create signature for a variety of cancers, indicating that the profile will help further establish molecular diagnosis, prognosis and therapy using miRNA.
Large high-throughput studies in patients revealed that miRNA profiling has the potential to classify tumors and predict patient outcome. miRNA deficiencies or excess have been correlated with a number of clinically important disease ranging from myocardial infarction to cancers. Multivariate regression analysis showed that the miRNA signature is independent from stage or histology and miRNA signature can predict patient survivals within cancer stages and histological subgroups of NSCLC patients. Beyond prognosis and prediction, miRNAs associated with neoplastic transformation may mediate pathophysiological mechanisms underlying tumorigenesis.
As mentioned, miRNA have been recently implicated in several carcinogenic process, where they can act either as oncogenes or as tumor suppressors. Therefore, therapeutic strategies based on modulation of miRNA activity hold great promise due to the ability of these small RNAs to potently influence cellular behavior.
More recently, it has been reported that serum and other body fluids contain sufficiently stable miRNA signature. Thus, the profiles of circulating miRNAs have been explored in a variety of studies aimed at the identification of novel non-invasive biomarkers. Particularly, concerning the functioning of circulating miRNA in the serum, miRNAs are currently extensively explored for their potential as non-invasive diagnostic tumor markers. Recent descriptions of miRNA in body fluids (such as blood, plasma, serum, and saliva) has suggested the potential for miRNA-based biomarkers in the detection of cardiovascular diseases such as myocardial damage, coronary artery disease, cancer and oral disease, indicating that miRNA may be excellent blood-based biomarker. These results, although preliminary, strongly suggest that a more comprehensive study of circulating miRNAs and their association with various physiopathological conditions may lead to another dimension in the discovery of biomarkers in the blood for many physiological and pathological conditions. Currently, there are clinical trials evaluating therapy based on miRNA inhibition or over-expression. Within the next several years, we will know if miRNA-based therapeutics, alone or in combination with other modalities, will be clinically useful treatments for various cancers and immune system disorders.
Bases on the body of knowledge from a a large numbers of studies, it can be concluded: a) miRNA-155 contributes to Treg cell development, 2) miRNA-dependent regulation is critical for preventing spontaneous inflammation and autoimmunity, 3) miRNA preserves the Treg cell functional program under inflammatory conditions, 4) miRNAs have a central role in monitoring the stability of differentiated Treg cell function in vivo and in the homeostasis of adaptive immune system, 5) Treg cell development involves Dicer-generated RNAs. Depleting miRNAs by eliminating Dicer, the RNAseIII enzyme that generates functional miRNAs, reduces Treg cell numbers and results in immune pathology. Dicer facilitates the development of Treg cells in thymus and the efficient induction of Foxp3 by TGF-β, and 6) miRNA can possibly be applied for diagnosis, treatment and prognosis for various diseases.
It is likely that this review is just the tip of the iceberg in terms of the involvement of regulatory RNAs in orchestrating immune response. It is perhaps not too optimistic to suggest that in the future, novel strategies for therapeutic intervention in immune related diseases could be based on manipulation of miRNA and Treg cells.
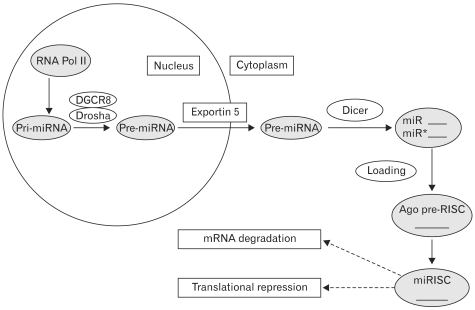
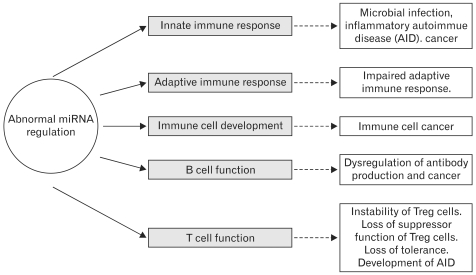
 XML Download
XML Download