

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Post-translational modifications (PTMs) are changes or alterations in a protein occurring after the completion of the translational process by ribosomes that are catalyzed by numerous enzymes. PTMs occur either when a functional group is covalently added to a protein, or during the proteolytic processing and folding processes. Protein PTMs play a key role in several physiological and cellular processes including cellular differentiation,1) protein degradation,2) signaling and regulatory processes,3) regulation of gene expression,4)5) and protein-protein interactions.6)7) PTMs act as a mechanism for the specification of proteins, through conformational changes that either minutely8) or largely9)10) change the overall tertiary structure of a protein.11) These modifications increase the variety and difference of proteins.12) Protein PTM dysfunction via external stimuli13)14)15) or aberrant signaling16)17) eventually leads to disease progression either through aberrant signaling or impaired PTM crosstalk.18)19) Non-native protein PTM leads to either biochemical dysfunction20) or a structural modification21) in the amino acid due to 'crosstalk.'12)22)
Mitochondria encompass 90% of the energy produced mainly by oxidative phosphorylation via electron transfer and adenosine triphosphate (ATP) synthase complexes.23) Subtle regulation of mitochondrial functions is mediated PTMs24) such as phosphorylation,25) acetylation,26) succinylation,27) and O-GlcNAcylation of mitochondrial proteins.28)29) Furthermore, since mitochondria are the central hubs of energy production, these networks are involved in various human pathological phenotypes; furthermore, mitochondrial proteomic dysfunction is directly associated with heart diseases.30)31) The availability of high-tech mass spectrometric techniques combined with advanced modified proteins/peptides by affinity chromatography methods has resulted in breakthroughs regarding the role of PTMs in cellular protein, particularly in mitochondrial proteins. Enzymes and stimuli involved in changes of PTMs are equally important; however, these issues are beyond the scope of this review. A better understanding of PTMs helps clinicians and researchers alike, but also facilitates development of targeted strategies for disease intervention.
Mitochondria, Heart Failure, and Post-Translational Modifications
According to the latest report by the World Health Organization, cardiovascular disease (CVD) is the leading cause of mortality in developed countries. Despite much progress in the advancement in the prevention, diagnosis, and management of CVD in the past decades,32)33)34)35) heart failure (HF) continues to be prevalent since current trends do not target direct cure in HF patients (except those with congenital heart diseases), but only decreases in the mortality rates.36)
Heart failure is a multifactorial clinical condition that is characterized by a dysfunction in the contractility of the myocardium, which results to the inability of the heart to provide enough blood for the metabolic needs of surrounding tissues.37) Increased preload and afterload, neurohormonal dysregulation, cardiac ischemia, and intrinsic abnormalities of the myocardium are common etiologic factors of HF. The gradual development of CVD to HF is a multicomponent and a multistep process, wherein cumulative acute cardiovascular injuries like myocardial ischemia/reperfusion (I/R) result in a chronic dysfunction.38)
HF progresses partly via changes in major signal transduction pathways,39) dysfunction in calcium homeostasis and energy fluxes,40) and alterations of the contractile apparatus in the heart.41) The underlying principles of bioenergetics is a key in further understanding its effects on CVD and HF (i.e., synthesized myocardial ATP transfer is required to maintain the excitationcontraction coupling, continuously supporting the optimal functioning of systolic and diastolic periods in the heart). Since cellular energetics and metabolism is heavily influenced and regulated by the mitochondria, there is undoubtedly a link between the heart and mitochondria function. Dysfunction in mitochondria such as oxidative damage,42) respiration impairment,43) and substrate utilization alterations have been reported in HF.44)
A frail heart undergoes complex changes in energy metabolism and substrate utilization due to mitochondrial dysfunction that are still unclear. These metabolic changes or remodeling occur when genes of interest trigger structural, functional, and electrical changes, which result in decreased cardiac function. One mechanism of this remodeling involves PTMs, which break down misfolded and damaged proteins in addition to proteins involved in contractile apparatus and hypertrophic gene expression. Likewise, proteins in respiratory chain and fat/glucose oxidation, which possess at least 1 reversible acetylation mark in complexes I, II, and V, and pyruvate dehydrogenase (PDH), and numerous acyl-CoA dehydrogenases, are involved.45)46) Regulation of metabolic enzymes within the mitochondria by acetylation implies that altered acetylation states within the mitochondria could play a role in the pathophysiology of heart failure.47)
Protein post-translational modifications are important in the study and analysis of disease progression such as those involving CVD, since the interplay between regulatory PTMs and the induced changes of the organelle dysfunction including mitochondria are potentially important factors in CVD progression.48) Several studies on the relationship of mitochondrial PTM and heart failure are reported.49)50) O'Rourke et al.48) in a study on heart mitochondria isolated from HF reported that cAMP-activated protein kinase might be involved in the increased protein phosphorylation during HF. Oxidative phosphorylation brought about by subunit-specific phosphorylation of complex IV regulates the incorporation or destabilization of the supercomplexes.51) In addition, growing evidence shows mitochondrial protein acetylation as a common mechanism in response to cardiac stress. Moreover, decreased nicotinamide adenine nucleotide (NAD+)/nicotinamide adenine nucleotide (reduced) (NADH) ratio and increased mitochondrial protein acetylation with increased sensitivity of mitochondrial permeability transition pore (mPTP) are associated during calcium sensitization. However, whether hyper-acetylation of a single protein or a select group of protein targets is the main contributor to increased sensitivity to cardiac stress remains to be determined.52)
The mitochondria plays a significant role in the regulation of cellular and physiological processes, hence it is important to study the associated proteins. We review the most common PTMs occurring in select mitochondrial proteins, as well as minor PTMs and the complex relationship with other PTMs that contribute to the progression of heart disease.
Post-Translational Modifications of Mitochondrial Proteins
Phosphorylation
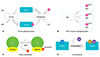
Signaling cascades are important in the study of acute cardiac pathologies, particularly in cases of I/R.12) Kinase activators and inhibitors are important mediators in pathological diseases.53) Phosphorylation is one of the well-studied PTMs that are responsible for altering a target protein's conformation that leads to either activation or inactivation of mitochondrial functions (Table 1 and Fig. 1A). Protein phosphorylation is a reversible PTM regulated by kinases and phosphatases, which is responsible for the phosphorylation and dephosphorylation of substrates, respectively.54) Protein kinases are key enzymes in modulating phosphate group transfer to serine, tyrosine, and threonine residues of the targeted proteins and thereby generate negatively charged side chains, which can either attract or repel target proteins in an external stimulus-dependent manner.55) Protein phosphatases on the other hand are signal transducing enzymes that dephosphorylate phosphoproteins.56)
Phosphorylation regulates the catalysis of numerous mitochondrial enzyme complexes by specific kinases associated with these complexes, making the process more efficient. One of the earliest studies regarding phosphorylation focuses on protein kinase capable of phosphorylating proteins in the rat liver mitochondria.53) Table 1 shows selected mitochondrial proteins from the Pagliarini and Dixon57) review on phosphoproteins found in the matrix, inner membrane, intermembrane space and outer membrane.
Pyruvate dehydrogenase complex (PDC), a kinase/phosphatasedependent regulatory cascade, is composed of 3 principle subunits: pyruvate dehydrogenase E1, dihydrolipoamide acetyltransferase E2, and dihydrolipoamide dehydrogenase E3. PDC links glycolysis to tricarboxylic acid cycle (TCA) cycle,58) catalyzing pyruvate conversion to acetyl CoA and is regulated by allosteric effectors and by reversible phosphorylation.59) E1 subunit complex phosphorylation at sites Ser264, 271, and 204 by pyruvate dehydrogenase kinase (PDK) leads to its inactivation,60)61) while dephosphorylation, via pyruvate dehydrogenase phosphatase results in its activation. Inhibition or phosphorylated state is brought about by high concentrations of immediate products such as acetyl CoA and NADH, and terminal products such as ATP levels. Thus, phosphorylation of PDC E1 by a phosphatase switches off the activity of the complex, thereby deactivating it (Fig. 1B).
Pyruvate dehydrogenase kinase (PDHK) exists mostly in organisms of eukaryotic lineages,62) and is notably absent only in organisms with reduced or absent mitochondria.61) The sequence of PDK bears significant similarity with histidine kinases,63)64) which are widely distributed sensory transducers in prokaryotes.65) PDHK is made up of 2 different subunits i.e., PDHK α-subunit with kinase activity on selective proteolytic cleavage and PDHK β-subunit, a regulatory subunit.66) Like phosphatase, kinase activities are activated by increased ratios of acetyl-CoA/CoA and NADH/NAD+67)68) and inhibited by elevated adenosine diphosphate (ADP) levels69) and by dichloroacetate70) in the mitochondrial matrix.
The inner mitochondrial membrane protein branched-chain α-ketoacid dehydrogenase (BCKD) complex, responsible for the oxidative decarboxylation of 3 branched amino acids (valine, leucine, and isoleucine), possesses a thiamine pyrophosphate-dependent branched-chain α-ketoacid decarboxylase (E1). Inactivation of this complex occurs via serine 193 phosphorylation through BCKD kinase (BCKDK).71) Branched chain amino acid levels are maintained during protein starvation by BCKDK-mediated phosphorylation and complex inhibition, a pathological characteristic of maple syrup urine disease that presents with severe neurological dysfunction.72)
End-stage heart failure may be partly due to reduced 3',5'-cyclic adenosine monophospate (cAMP)-dependent phosphorylation in various oxidative phosphorylation (OXPHOS) subunits.73) cAMPdependent phosphorylation regulates OXPHOS activity by elevating cAMP levels, which leads to increased nicotinamide adenine dinucleotide dehydrogenase (ubiquinone) Fe-S protein 4 (NDUFS4) phosphorylation complex I (CI) activity by twofold.74) In line with this, CI reportedly undergoes cAMP-responsive phosphorylation at the 10 and 18-kDa subunits.75) Mass spectrometry technique also shows that purified CI possesses phosphorylation sites at the 42-kDa subunit in addition to the B16.6, B14.5a, and B14.5b subunits of CI.76) Complex IV (CIV) subunit phosphorylation is considered not only beneficial, but critical for CIV activity in healthy and pathological cardiac mitochondria. Hypoxia and ischemia increase protein kinase A-dependent phosphorylation of IVi1 and Vb subunits of CIV,77) which are associated with lower CIV activity and increased reactive oxygen species (ROS) production.78)
Currently, phosphorylation is known to regulate proteins in the different mitochondrial compartments in a very specific manner, and its regulation leads to alterations that either directly or indirectly cause heart failure. However, the kinases and phosphatases involved during mitochondrial protein phosphorylation, including its subunits are not fully understood. In addition, the phosphorylation events regulating mitochondrial functions or dysfunctions in vivo remain to be confirmed.
Acetylation
Acetylation occurs when an acetyl group is introduced into a compound, wherein the hydrogen atom of a hydroxyl group is exchanged with an acetyl group yielding acetate.79) Studies have suggested that non-histone-acetylases and deacetylases are both involved in cardiac remodeling (Table 2 and Fig. 1C).
Sirtuins (SIRTs) are NAD+-dependent protein deacetylases that play key roles in regulation of mammalian metabolism. Cells have 7 different SIRTs, 3 of which are localized in the mitochondria: SIRT3, SIRT4, and SIRT5.80) When SIRT3 is abolished, mitochondrial proteins become hyperacetylated and exhibit altered function that eventually leads to mitochondrial dysfunction.81) SIRT3 targets many enzymes, which is suggestive of its role in heart failure. For example, mitochondrial protein acetyl-CoA synthetase 2 (AceCS2) activities are directly regulated by SIRT3 in an NAD+-dependent manner.82)83) SIRT3 in neonatal rat ventricular myocytes (NRVMs) deacetylates nuclear protein Ku70, preventing mitochondrial translocation of Bax and enhancing H2O2 tolerance.84) SIRT3 also deacetylates FOXO3a responsible for increasing manganesedependent superoxide dismutase (MnSOD, SOD2) expression, which in turn diminishes mitochondrial superoxide.85) A new study has shown the role of SIRT3 in fatty acid oxidation (FAO), wherein it deacetylates Lys42 of the 8 acetylation site on long-chain acyl-CoA dehydrogenase (LCAD) leading to FAO pathway activation.86) Aside from AceCS2 and LCAD, acetylation also regulates other mitochondrial components such as malate dehydrogenase87) and isocitrate dehydrogenase88) in the TCA cycle. Mitochondrial malate dehydrogenase (MDH2) becomes acetylated at Lys185, Lys301, Lys307 and Lys314 and Lys314, resulting in increased activity (rapid reduction of oxaloacetate to malate when coupled with high NADH) during the hyperacetylated MDH2 state,89) thereby increasing gluconeogenesis and hyperglycemia risks.87) On the other hand, SIRT4 and SIRT5-deficient mice do not show any significant changes in their acetylation status,90) and thus both SIRT4 and SIRT5 have limited deacetylase activities.91)
S-nitrosylation
S-nitrosylation is the specific attachment of nitric oxide (NO)-related species to a thiol group to form S-nitrosothiol, which also serves as a cellular signal due to its similar nature with phosphorylation.92) The actions of denitroylases are thought to be similar with those of the phosphatases in kinase signaling; in addition, they are considered as major regulators of the cytosolic and mitochondrial thioredoxin reductases.93)94)
In mitochondria, regulation of oxidative phosphorylation and glycolysis by NO-mediated protein S-nitrosylation is important in both physiologic and pathophysiologic conditions (Fig. 1D). S-nitrosylation of mitochondrial aldehyde dehydrogenase 2 family, involved in NAD+-dependent oxidation of the different aldehydes produced during intermediary metabolism, leads to reversible inhibition.95) Creatine kinase, responsible for conversion of creatine, uses ATP to produce phosphocreatine and ADP; it is inhibited by S-nitrosoglutathione (GSNO) dose-dependently via transnitrosylation96) and reversibly regulated through S-nitrosylation of Cys283 in adult rat ventricular myocytes.97) Another study using isolated rat heart mitochondria as a model shows inhibition of complex I by the S-nitrosylation of the 75-kDa subunit through the exogenous addition of GSNO. In an endothelial cell model, the inhibition of mitochondrial complex IV/cytochrome c oxidase possibly occurs by S-nitrosylation at Cys196 and Cys200, which are both active residues.98) However, the exact mechanism of this PTM in vivo, briefly described as the addition of NO to reactive cysteines, still remains largely unknown.99)100)
Cross-talk with various protein post-translational modifications
The respective actions of each PTM are clearly essential modulators of protein structure-function relationships. The numerous PTMs are potentially related, forming networks, as evident in different biological systems.101) Three criteria are suggested in crosstalk of PTMs: 1) similar site competition; 2) modification that facilitates conformational change in the second site accessibility that allows another PTM to occur, and; 3) direct alteration of the modifying enzyme of the other PTM.102) One of the earliest studies concerning potential PTM crosstalk focuses on the potential sites of O-linked β-N-acetylglucosamine (O-GlcNAc), which is modification of serine or threonine hydroxyl moieties by β-N-acetylglucosamine, or phosphorylation modification, which both target serine and threonine dynamically and transiently in nature.28) However, a recent study shows that the location of the O-GlcNAcylation machinery within the cell partially dictates its function. The regulation of O-GlcNAcylation through subcellular redistribution of OGT/OGA and functional consequences that have immediate therapeutic potential to improve cardiac contractility posits a new concept in PTM cross-talk.103) Furthermore, PTM sumoylation is the covalent protein modification by addition of ubiquitin-like polypeptides. Crosstalk between sumoylation and phosphorylation is also suggested, since the small ubiquitin-like modifier (SUMO) attachment lysine site is located 4 sites from a phosphorylated serine in numerous sumoylated proteins.104)105)
Phosphorylation and lysine acetylation are likewise involved in crosstalk.23) For example, adenosine monophosphate-activated protein kinase and the SIRT family. A study shows that there is an approximately 80% overlap between the interacting sites of mitochondrial lysine acetylation and succinylation; for instance, approximately 25% of known SIRT5 target sites were also modified by SIRT3.106) These findings indicate the possible cooperation between proteins in order to maintain balance in the mitochondria.
Crosstalk also signals degradation, as shown in I/R injury where myosin light chain 2 is reduced between a deamidated asparagine and a phosphorylated serine, exhibiting 3 PTMs occurring within 2 amino acids. Similarly, there is a complicated relationship between S-glutathionylation and major PTMs. S-glutathionylation, in which protein cysteine residues are modified during glutathione addition, has 2 main mitochondrial roles i.e., oxidant stress defense and redox signaling. During the oxidation of the mitochondrial glutathione, the cysteine-rich 75-kDa subunit of complex I becomes the main target.107) Glutathionylation/oxidation acts as a buffer against ROS under these conditions, keeping protein thiols away from the gradual oxidation to sulfinic acid and sulfonic acid, which might lead to irreversible protein dysfunction (Fig. 1).108)109) Another study108) shows that glutathionylation of Complex I (CI) due to diamide-induced glutathion depletion inhibits CI activity; however, ROS levels remain unchanged during nitrosylation on the same subunit. CI undergoes nitrosylation and glutathionylation, hence, mitochondrial complex II (CII) is a protein that persistently undergoes glutathionylation. In an I/R model induced by coronary ligation, the 70-kDa subunit of CII undergoes markedly reduced glutathionylation that is related with the loss of electron transfer activity. Thus, glutathionylation likely plays a key role in the maintenance of CII function.110)
Limitations of mitochondrial protein post-translational modification studies
With the technological advancements of recent years, hundreds of mitochondrial proteins are identified through mass spectrometry-based proteomics. This has led to a completely novel way of understanding CVD. Extensive validation of PTM sites on mitochondrial proteins and the correlation with protein function remains to be done. A full understanding of the intricate web of post-translational signaling and regulation in the mitochondria, as well as the identification of target proteins, should be the next step before further development of strategies to combat CVD. However, integrative approaches including computational biology, protein arrays, and biochemical analyses will quickly advance the progress of studies. Hofer and Wenz111) raised several questions whether PTMs are regulatory in nature given the small percentage of target proteins that are modified during the process; and if so, the regulatory mechanisms involved need to be elucidated. In addition, it is not clear whether all regulatory PTMs are beneficial to the continued functioning of the system, or detrimental. Moreover, questions regarding tissue-specificity and time-dependence should be considered during experimentation. Addressing these issues will greatly aid in understanding disease mechanisms and suggest targeted strategies for disease intervention.
A Clinical Perspective of Mitochondrial Protein Post-Translational Modification for Combating Cardiovascular Disease
Current therapies do not directly address the treatment of CVD, but rather, are aimed at slowing its progress to HF. For example, statins that slow the formation of atherosclerotic lesions,112) antianginals targeting ischemic tissues,113) and antiplatelet or anticoagulation agents which hinder the formation of a clot.113) Cardiac ischemia can at times be predicted, such as in the cases of cardiac surgery or balloon inflation employed in percutaneous coronary intervention.114) More recently, preconditioning (PC) of the heart has emerged in clinical practice,115) wherein the heart is subjected to short intermittent I/R cycles before index ischemia that reduces the infarct size and improves postischemic function.116) Targeting the MPTP is a new trend with more promising effect.
MPTP opening is a key factor that drives necrotic cell death in I/R injury.117) The key components of MPTP are still unclear, but transgenic animal models show that adenine nucleotide translocase as the phosphate carrier can regulate the pore opening.118) Cyclophilin D119) and voltage-dependent anion channel (VDAC)120) are associated with MPTP but their roles remain unclear. Other proteins related with MPTP include hexokinase II,121) which serves as a connection between the pore and cellular metabolism and mitochondrial translocator protein, which communicates with VDAC.122)
PC can act on MPTP either directly on the MPTP to inhibit its opening123) and/or by reducing calcium or ROS levels that trigger MPTP.117) Considering that cardioprotective signaling pathways are activated through direct MPTP inhibition, these signaling pathways are capable of modifying MPTP components. Thus, we can expect that cardioprotective signaling leads to mitochondrial protein PTM.
The importance of epigenetics in gene regulatory mechanisms leading to cardiovascular complications have also been widely studied (particularly histone and DNA modifications) as a therapeutic target for CVD. Factors such as diet, environmental changes, and activity affect gene expression in an entity and its offspring via epigenetics, without affecting the genomic sequence. A clinical study124) conducted by the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complication (DCCT/EDIC), show that patients on constant intensive therapy have lower cardiovascular complications, as compared to those who are first on conventional therapy and then switched to intensive therapy. This supports the evidence that a history of hyperglycemia possibly leads to long-lasting molecular changes that ultimately puts the patients on the fast-track to the development and progression of CVD. Thus, PTM-mediated regulation of the cardiac proteome can serve as a foundation laid early in life or transmitted from the parental units.125)
HF is a multifactorial syndrome that progresses largely due to myocardial dysfunction brought about by mitochondrial modifications. Therefore, it is important to understand mitochondrial cytopathy in HF as a potential foundation for therapeutic strategies to maintain mitochondrial integrity, enhance substrate metabolism, protect and reduce oxidative stress in the environment, and improve the myocardial contractility.126) These changes are undoubtedly related to the modification of mitochondrial proteins through various PTMs. Some novel mitochondrial targets of phosphorylation, S-nitrosylation, and acetylation are elucidated, most of which are linked to single protein activity or total mitochondrial function. Collectively, the understanding of mitochondrial PTMs will provide an insight into controlling mitochondria-related conditions such as HF, but more systemic and long-term research is needed.


 XML Download
XML Download