

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Sensitizing epidermal growth factor receptor (EGFR) mutation such as G719X, deletion on exon 19, L858R or L861X in non-small cell lung cancer (NSCLC) is a strong predictive marker for EGFR-tyrosine kinase inhibitors (EGFR-TKIs)1,2. However, despite initial dramatic response in patients harboring such EGFR mutations, acquired resistance eventually develops in most of them3,4. Through enormous efforts to identify and understand resistance mechanisms, several causes and overcoming strategies have been revealed. Among them, secondary threonine-to-methionine mutation at codon 790 in EGFR (T790M) is the most common mechanism representing almost 50% of them5,6. More potent irreversible EGFR-TKIs and mutant selective EGFR inhibitors have been actively investigated for the possibility of therapeutic options to overcome T790M-mediated resistance7-9. Bypass signals by met proto-oncogene (MET) amplification or AXL receptor tyrosine kinase (AXL) overexpression also have been suggested as other possible causes leading to resistance. Several MET and AXL inhibitors are currently under clinical trials or development because combined treatment with EGFR-TKIs showed excellent response in experimental models10,11.
In 2011, Sequist et al.12 reported the frequency of observed EGFR-TKI resistance mechanisms by analysis of patients undergoing post-resistance biopsy at Massachusetts General Hospital. The detection rate of T790M (49%) was not different, while that of MET amplification (5%) was much lower in this cohort, compared to those of previous studies10,13. Interestingly, small cell lung cancer (SCLC) transformation from NSCLC was found in 5 patients (14%) although its original EGFR mutation was maintained in resistant tumors, and 3 of them showed the response to chemotherapy for SCLC. Considering that there have been several similar case reports14, SCLC transformation would be one of the resistance mechanisms for EGFR-TKI therapy. However, it is not clear whether combined SCLC cells selected by EGFR-TKI treatment were initially present in un-biopsied parts of tumor or real SCLC transformation from NSCLC occurred in tumors of those patients. In this study, we investigated whether it would be possible to find the phenomenon of SCLC transformation or neuroendocrine (NE) differentiation in EGFR-TKI-resistant cell line models, and that could affect the response to conventional chemotherapeutic drugs for SCLC treatment.
Materials and Methods
1. Cell culture and reagents
The A549 and HCC827 cells were purchased from the American Type Culture Collection (Rockville, MD, USA). The PC-9 cell line was a kind gift from Dr. Kazuto Nishio (National Cancer Center Hospital, Tokyo, Japan). All cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum, 100 U/mL penicillin and 100 µg/mL streptomycin (Invitrogen) at 37℃ in an atmosphere of 5% CO2. Gefitinib, erlotinib (reversible EGFR-TKIs) and ZD6474 (an EGFR and vascular endothelial growth factor receptor inhibitor) were purchased from Selleck Chemicals (Houston, TX, USA). CL-387,785 (an irreversible EGFR-TKI) was purchased from Calbiochem (San Diego, CA, USA). The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution, cAMP, 3-isobutyl-1-methylxanthine (IBMX), cisplatin and etoposide were all purchased from Sigma (St. Louis, MO, USA).
2. Establishment of the EGFR-TKIs-resistant cell lines
The A549/GR, PC-9/GR, PC-9/ER, and HCC827/CLR were established in our previous researches15-17. The PC-9/CLR and PC-9/ZDR cells were established by using the previously described method15. Briefly, PC-9 cells were exposed to 10 nmol/L of CL-387,785 or ZD6474 for 72 hours in media containing 10% fetal bovine serum. Then, they were washed and cultured in drug-free media until surviving cells were 80% confluent. These cells were re-exposed to increasing concentrations of CL-387,785 or ZD6474. Cells which could grow in the 100 nmol/L CL-387,785 or ZD6474 were obtained 6 months after initial exposure. The surviving cells were cloned, then, the CL-387,785 and ZD6474-resistant cell lines were designated as PC-9/CLR and PC-9/ZDR, respectively. For all in vitro studies, resistant cells were cultured in drug-free media for at least 1 week to eliminate CL-387,785 and ZD6474.
3. MTT assay
Briefly, the cells were seeded onto 96-well plates overnight. Each of drugs was added to them in a dose-dependent manner, then, the cells were incubated for 72 hours. The viability of cells was determined by using the MTT assay in accordance with the method described by Carmichael et al.18.
4. Western blot analysis
The cell lysates were prepared by using the previously described method19. The membrane was probed with antibodies against CD56, synaptophysin (SYP), chromogranin (Chr-A) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-Rb, Rb, p16, poly(ADP-ribose) polymerase (PARP) and β-actin (Cell Signaling Technology, Beverly, MA, USA) as primary antibodies, and then the membrane was treated with horseradish-peroxidase-conjugated secondary antibody. The membrane was developed using an ECL kit (Amersham Biosciences, Piscataway, NJ, USA).
5. Immunohistochemistry analysis
The cells were grown in a chamber slide (Nalge Nunc International, Naperville, IL, USA). They stained with H&E visualized with a Nikon light microscope (Nikon, Inc., Melville, NY, USA) that had the capability of digital photography. For immunohistochemical analysis, the paraffin sections (4 µm thick) were deparaffinized with xylene, rinsed thoroughly with ethanol and then soaked in 0.03% hydrogen peroxide in methanol to inactivate the endogenous peroxidase activity. The sections were incubated with either 10% goat serum or 10% rabbit serum, and then they were covered with the primary antibodies, washed with phosphate-buffered saline and processed further with using a DAKO EnVision kit (DAKO, Los Angeles, CA, USA), as directed by the manufacturer. The color was developed with 3,3'-diaminobenzindine (DAB) that contained 0.3% H2O2. Primary antibodies against CD56 and SYP were used.
6. Induction of NE differentiation by cAMP/IBMX
To induce NE differentiation by the previously described method20, A549 cells were seeded at 50% confluency in a chamber slide. The cells were treated with 0.5 mmol/L cAMP/IBMX combinations for 72 hours, and then they were fixed for 1 hour with methanol. Immunocytochemical staining of the cells was performed by the same procedure used for the paraffin sections.
Results
1. NE differentiation only in A549/GR cells leading to increased chemosensitivity
We investigated the change of NE markers in lung cancer cells which acquired resistance to EGFR-TKIs by performing western blotting and immunocytochemistry. PC-9/CLR and PC-9/ZDR cells were cloned with continuously exposed to increasing concentrations of CL-387,785 (IC50: <1 nM in the PC-9 cells and >100 nM in the PC-9/CLR; data not shown) or ZD6474 (IC50: 50 nM in the PC-9 cells and 500 nM in the PC-9/ZDR; data not shown), and other EGFR-TKI-resistant cells were established in previous studies15-17. The expression of CD56 was increased in both the A549/GR and PC-9/ZDR cells (Figure 1A). However, the enhanced expression of SYP, Rb and p16 were found only in the A549/GR cells (Figure 1B, C). As morphological changes, the cluster of A549/GR cells became more compact and their nuclear/cytoplasmic ratios were increased. These cells were also more sensitive to etoposide or cisplatin than parental A549 cells (Figure 1D, E). These results suggest that NE differentiation occurred in A549/GR cells.
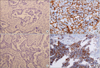
2. Increased CD56 and SYP expression in a rebiopsied tumor after development of resistance to erlotinib
Rebiopsy of regrowing tumor during treatment with erlotinib was performed in a patient with adenocarcinoma who had initially responded well to erlotinib. A deletion mutation on exon 19 of EGFR gene was noted in the tissues of the regrowing tumor as well as in those of the original tumor. Interestingly, the expression of CD56 and SYP was increased after acquiring resistance (Figure 2), which suggests that NE differentiation occurred in this patient.
3. Induced NE differentiation by cAMP and IBMX with increasing chemosensitivity
A previous study demonstrated that combined cAMP and IBMX treatment led to NE differentiation in NSCLC cells20. We induced NE differentiation of A549 cells by treating them with the same method. When the cells were co-treated with cAMP and IBMX, they became scattered and extended with a slender shape (Figure 3A). The expression of SYP was increased on the western blots and immunocytochemistry while that of Chr-A was slightly increased. However, CD56 expression was not observed (Figure 3B, C). Transformed cells obtained the increased sensitivity to etoposide and cisplatin compared to parental cells (Figure 3D).
4. Increased PARP expression in A549/GR cells without affecting to the response to chemotherapeutic drugs by its inhibition
Recently, overexpression of PARP1 and enhancer of zeste homolog 2 (EZH2) has been indentified in an integrated analysis of multiple proteins involved in intracellular signaling pathway in SCLC cell lines21. They showed that PARP1 was highly expressed at the mRNA and protein levels in SCLCs, and then SCLC was significantly more sensitive to PARP inhibitors than NSCLCs. Thus, they suggested that PARP inhibition downregulated the key components of DNA repair machinery and enhanced the efficacy of chemotherapy. Consistent with this, PARP1 expression was increased in A549/GR cells (Figure 4A). However, the response to AZD2281, a PARP inhibitor, was not different between A549 and A549/GR cells (Figure 4B). In addition, PARP inhibition by AZD2281 did not affect the response to etoposide and cisplatin (Figure 4C).
Discussion
This is the first study showing that NE differentiation from NSCLC cells occurred during acquisition of resistance to EGFR-TKI to date. In general, EGFR-TKIs don't seem to be effective in SCLC although some early experimental studies suggest their efficacy in cell line models22. Only 2 of 19 patients with SCLC showed stable disease while 17 patients had progressive disease by gefitinib in a phase II study23. However, the benefit from gefitinib therapy could be obtained if SCLC would have a sensitizing EGFR mutation although it is very rare. A Japanese never-smoker patient with SCLC responded to gefitinib which was initiated at patient's request24. Deletion mutation on exon 19 (delE746-A750) was found by direct sequencing in her tumor tissue. Zakowski et al.25 also reported a female American case of SCLC with deletion mutation on exon 19. Erlotinib led to partial response and the response duration was 18 months.
However, it is too early to conclude that the EGFR mutation in SCLC is a predictive marker for EGFR-TKIs because SCLC transformation harboring EGFR mutation was found in resistant tumor samples after EGFR-TKI therapy. If the assertion that SCLC transformation is one of the resistant mechanisms to EGFR-TKI would be true, what's the difference of EGFR mutations between EGFR-TKI-responsive and -resistant SCLC? This question leads us to consider the possibility that SCLC transformation or NE differentiation could be an accompanying phenomenon occurring during acquisition of resistance, not the main cause of resistance. We expect it could be elucidated through further studies in near future.
Approximately 15% of NSCLCs have NE features26 and they have been suggested to exhibit the biological characteristics similar to SCLC such as early metastasis, initial responsiveness to chemotherapeutic drugs and expression of NE markers including neuron-specific enolase, L-dopa decarboxylase, Chr-A, SYP, and cytoplasmic dense core granules. However, there have been conflicting results regarding chemosensitivity of NSCLC with NE features. Some revealed that NE differentiation led to chemosensitivity27,28 while others failed to demonstrate any correlation with response to drugs29-31. Probably, this might be caused by discrepancies on how to define NE differentiation, how to interpret the results of immunohistochemical staining and what kinds of antibodies should be used. In our study, regardless of whether it exerts a significant role in resistance to EGFR-TKI or not, NE differentiation made transformed cells more susceptible to chemotherapeutic drugs such as etoposide and cisplatin which are currently being used for treatment of SCLC, suggesting that these drugs could be the next therapeutic option.
The case reported by Morinaga raised another question whether SCLC found in resistant samples arose from the transformation of adenocarcinoma or the selection of pre-existing minor clone14. The first and third biopsies after treatment of irinotecan and cisplatin revealed adenocarcinoma, while the second biopsy at the same site in the mass in progress during EGFR-TKI therapy showed SCLC in that report. Although the biopsies were done at the same site, it seems to be very hard to prove the reality of transformation because small pieces of tissues procured by needle biopsy cannot represent the whole nature of cancer. However, our study performed with cell lines firstly demonstrated that NE differentiation can occur during EGFR-TKI treatment.
Recently, Byers et al. investigated molecular characteristics of SCLC using an integrative proteomic and transcriptomic analysis21. They found that the expression of PARP1, a DNA repair protein and E2F1, co-activator, was highly increased both at the mRNA and protein levels in SCLC, and SCLC growth was inhibited by PARP1 knockdown. In our study, PARP1 expression was also increased in A549 cells with NE differentiation compared to parental cells. However, there was no difference in the response of A549/GR cells to a PARP1 inhibitor compared to parental cells. Further, the combination of AZD2281 with etoposide or cisplatin could not enhance the growth-inhibitory effect in A549/GR cells. These are clearly contrasted with results from Byers et al. suggesting that although A549/GR cells were differentiated to possess some NE features, they would not acquire the whole characteristics of SCLC. However, further studies would be required for the response to EGFR-TKI treatment of other lung cancers with NE differentiation such as large cell carcinoma or carcinoid tumor.
Although we proved that NE differentiation can occur during EGFR-TKI treatment, not by selection of pre-existing cells, established A549/GR does not seem to be clinically appropriate model because A549 cells do not harbor EGFR mutations. Nevertheless, we had a similar experience in epithelial to mesenchymal transition (EMT) by EGFR-TKI. This phenomenon was also firstly found in A549 cells with acquired resistance to gefitinib15, which was also not appropriate, it led to the discovery of EMT in EGFR-mutant lung cancer cells16 and clinical samples12,17 later.
In summary, NE differentiation occurred during acquisition of resistance to EGFR-TKI leading to increased chemosensitivity suggesting therapeutic drugs for SCLC would be useful. However, it should be more clarified whether NE differentiation is the real main cause of EGFR-TKI resistance.



 XML Download
XML Download